体外诱导对氨基水杨酸高浓度耐药结核分枝杆菌及其突变位点研究
2023-02-10余美玲张晨晨魏文静赵雨川卓文基郑磊
余美玲 张晨晨 魏文静 赵雨川 卓文基 郑磊
对氨基水杨酸(para-aminosalicylic acid,PAS)是目前用于治疗耐多药结核病的二线抗结核药物[1]。尽管PAS用于结核病治疗已超过70年,但对其作用机制尚未完全了解[2]。目前,普遍认为结核分枝杆菌(Mycobacteriumtuberculosis,MTB)对PAS耐药主要与某些基因突变有关,包括thyA、folC和ribD等[3]。研究发现,thyA基因编码胸苷酸合成酶,其缺失可导致MTB对PAS的耐药性[4],thyA基因的突变也存在于对PAS耐药的MTB临床分离株中[5]。作为对氨基苯甲酸(para-amino benzoic acid,PABA)的类似物,PAS与PABA竞争二氢叶酸合成酶 (DHPS),从而干扰叶酸合成[6-8]。此外,也有研究显示,编码二氢叶酸合成酶的folC基因的各种错义突变和ribD的过表达可使MTB对PAS产生耐药性[9]。然而,folC突变仅在34.8%的MTB临床耐药分离株中被检测到,而thyA和ribD突变分别在26.0%和5.8%的MTB临床分离株中被检测到[5, 7]。
目前,MTB耐药研究多采用临床分离的药物敏感和耐药菌株进行对比分析[10-11],难以消除不同菌株的背景差异,无法得知MTB耐药进化轨迹,亦无法准确获得MTB耐药变化的始动因素及引起耐多药的关键因素和共同因素。笔者采用世界卫生组织(World Health Organization,WHO)推荐的改良罗氏固体培养法及药物敏感性试验(简称“药敏试验”)方法,制备不同药物浓度梯度的PAS培养基,通过连续传代和持续诱导的方式,将MTB标准菌株H37Rv成功诱导成标准耐PAS菌株(WHO推荐的耐药浓度)及高水平耐药菌株,收集并保存每一代诱导菌株,建立动态耐药菌株遗传进化模型。用微孔板法检测上述诱导菌株的最低抑菌浓度(minimum inhibitory concentration,MIC)及交叉耐药情况,筛选耐药水平相似的临床分离株。通过对PAS耐药的实验室诱导MTB菌株和临床分离株进行全基因组测序,研究PAS潜在的作用和MTB对PAS的耐药新机制。另外,基于临床用药实际情况,构建连续、动态的耐PAS的MTB菌株模型,以期为MTB对PAS耐药研究和临床实践提供理想的生物模型。
材料和方法
一、研究材料
1.菌株:本研究所用MTB临床分离株来自广东省结核病控制中心生物样本信息库,菌株的PAS药敏信息齐全;MTB标准株H37Rv为本实验室保存。本实验室为省级结核病参比实验室,设有MTB菌株暂存库,按相关要求存储菌株;已在当地卫生健康委员会备案涉及病原微生物为MTB的二级病原微生物实验室[备案编号为:BSL-2(2020)44010600068]。少量活菌操作时在加强型二级生物安全实验室进行,实验人员进行三级防护。
2.试剂与培养基:实验所用Middlebrook 7H9培养基、Middlebrook 7H10培养基和OADC增菌剂均购买于美国BD公司,PAS购买于美国Sigma公司。MTB药敏试验试剂盒购自珠海贝索生物技术有限公司(货号BC8001)。
改良罗氏固体培养基由本实验室配制,配方基础成分(g/600 ml):磷酸二氢钾2.4 g、硫酸镁0.24 g、柠檬酸镁0.6 g、L-谷氨酸钠7.2 g、孔雀绿0.4 g、马铃薯淀粉30.0 g。基础成分购自北京索莱宝科技有限公司。配制方法:称取基础成分49.84 g,并吸取甘油12 ml,加热搅拌溶解于600 ml蒸馏水中,煮沸5~10 min,121 ℃高压灭菌15 min取出。待冷却至55 ℃左右时,以无菌操作方法加入无菌搅匀的全蛋液1000 ml,混匀(避免产生气泡),分装18 mm×180 mm 试管,每管7 ml,置于成长斜面,用85~90 ℃流动蒸汽加热1~1.5 h后,取出放冷备用。含PAS的罗氏固体培养基在此基础上,配制时分别加入不同浓度的PAS。WHO推荐的MTB对PAS耐药临界浓度为1.0 mg/L[12]。
二、研究方法
1.体外诱导耐PAS的MTB菌株:采用WHO推荐的改良罗氏固体药敏试验,参照MTB传统药敏试验培养基中PAS浓度,配制一系列的药物浓度梯度,依次将各药物稀释1倍,稀释4个梯度,即得到不同的药物浓度20、2-1、2-2、2-3、2-4。挑取H37Rv单克隆菌株接种于中性罗氏培养基上进行扩大培养,该菌株视为原始初代菌株P0代。随后将该菌株分别接种在不同浓度的PAS培养基上,37 ℃培养4周,其间记录各浓度培养基上菌株的生长状况;4周后挑选生长状态较好的一组进行下一轮传代,所有药物浓度梯度提高1倍。依此类推,筛选出每一代生长良好的菌株,连续传代,直至筛选的阳性克隆株的MIC值达到WHO推荐的PAS耐药临界浓度(1.0 mg/L)。另外,为进一步探索MTB获得高水平PAS耐药的机制,继续给予上述诱导成功的标准菌株21、22、23倍药物浓度压力刺激,保留每一代阳性菌株,从而建立每种药物连续诱导的MTB耐药动态变化模型株,有助于分析MTB药物压力下的遗传进化和耐药机制。
2.筛选耐药水平相似的临床分离株:从本中心的生物样本库中筛选耐药水平相似的PAS耐药临床分离株和PAS敏感临床分离株,并用液体微孔板法进行药敏试验复核。
3.MTB菌株药敏试验:用液体微孔板法检测本研究所用MTB菌株对14种抗结核药物[异烟肼(INH)0.025~4 mg/L、链霉素(Sm)0.25~40 mg/L、乙胺丁醇(EMB)0.31~20 mg/L、氧氟沙星(Ofx)0.25~16 mg/L、莫西沙星(Mfx)0.06~4 mg/L、阿米卡星(Am)0.25~16 mg/L、卡那霉素(Km)0.62~20 mg/L、卷曲霉素(Cm)0.5~16 mg/L、丙硫异烟胺(Pto)0.62~20 mg/L、PAS 0.5~16 mg/L、利福平(RFP)0.25~8 mg/L、利福布汀(Rfb)0.12~4 mg/L、左氧氟沙星(Lfx)0.25~16 mg/L和吡嗪酰胺(PZA)50~900 mg/L]的MIC及交叉耐药情况。
4.MTB基因组DNA提取及测序:取400 μl上述灭活的诱导菌株、临床分离株及作为对照组的H37Rv菌株的菌液进行超声分散。运用十六烷基三甲基溴化铵(hexadecyltrimethy ammonium bromide,CTAB)法提取MTB全基因组,并用 NanoDrop 测定浓度,然后用0.8%~1%琼脂糖凝胶电泳检测样品质量。使用Agilent 5400对DNA纯度、浓度、完整性进行检测。DNA样品检测合格后,使用Covaris超声波破碎仪随机打断,再经末端修复、加A尾、加测序接头、纯化、PCR扩增等步骤完成整个文库制备工作。文库构建完成后,先使用Qubit 2.0进行初步定量,稀释文库,随后使用Agilent 2100对文库的插入片段进行检测。插入片段大小符合预期后,使用实时荧光定量PCR方法对文库的有效浓度进行准确定量,以保证文库质量。文库检测合格后,按照有效浓度及目标下机数据量的需求将不同文库pooling至flowcell,cBOT成簇后使用美国Illumina公司高通量测序平台NovaSeq 6000进行测序。测序数据已上传Sequence Read Archive (SRA)公共数据库,BioProject编号为PRJNA883518和PRJNA883540。
三、数据分析
1.MTB突变分析:(1)低质量序列过滤、接头(adapter)序列去除:测序得到的原始数据(Raw Data)会存在一定比例的低质量数据,为了保证后续信息分析结果的准确可靠,采用Fastq(0.20.0)质控软件对原始数据进行质控,得到有效数据(Clean Data)。(2)参考序列比对:从各样品质控后的有效数据出发,将有效数据比对到MTB参考基因组H37Rv(NC_000962.3)进行测序深度和覆盖度的统计(BWA 0.7.17)。(3)变异检测:通过与参考基因组H37Rv进行比对结果(Bam文件),鉴定突变位点(Freebayes 1.3.2),进行变异检测,对突变进行注释(同义、错义、无意、移码突变等),获得目标基因组对于参考基因组的单核苷酸多态性(single nucleotide polymorphism,SNP)及插入缺失(insertion-deletion,InDel)等一系列变异信息(SnpEff 4.3t)。
2.耐药分析:从突变分析中生成的Bam文件提取各耐药位点的序列比对信息,分析各耐药位点的突变,根据耐药位点数据库(https://github.com/jodyphelan/tbdb)中突变和耐药关系,预测耐药表型。
结 果
一、诱导耐PAS的MTB菌株
采用WHO推荐的改良罗氏固体药敏试验,在体外诱导产生不同耐药水平的PAS耐药菌株,期间保留每一代阳性菌株,建立耐PAS的MTB耐药动态变化模型(图1)。诱导过程中,在第三代,MTB菌株(P3)对PAS出现耐药性,但菌株生长状态较差,连续培养至第七代(P7),菌落形态与无药对照组相似。然后继续培养三代,稳定耐药性状,至此获得PAS临界耐药的MTB菌株。为进一步探索MTB获得高水平PAS耐药的机制,继续给予上述诱导成功的标准菌株2、4、8倍药物浓度压力,并分别获得对应的耐PAS菌株(P15、P20和P26)。
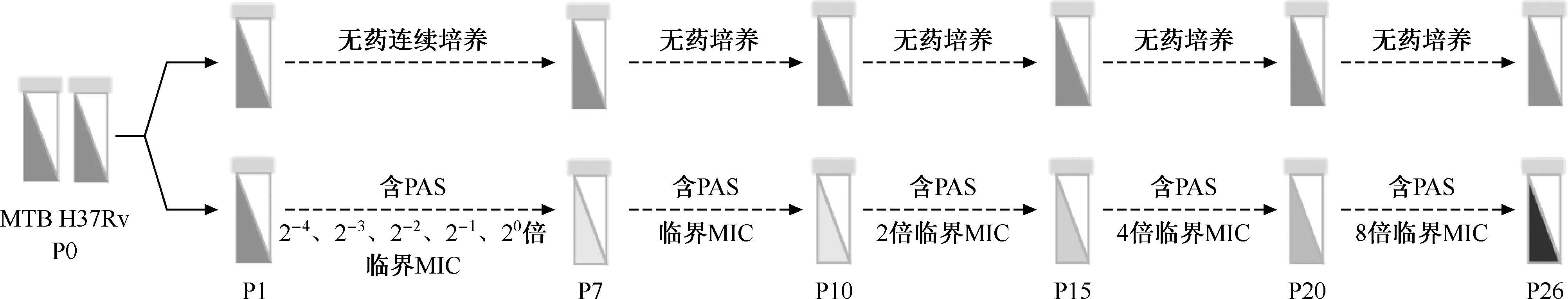
注 MTB:结核分枝杆菌;PAS:对氨基水杨酸;MIC:最低抑菌浓度图1 体外诱导对氨基水杨酸耐药结核分枝杆菌菌株示意图
二、MTB药敏试验验证耐药表型
用液体微孔板法检测上述诱导菌株对14种抗结核药物的MIC及交叉耐药情况。结果显示,在诱导培养至第三代时即出现PAS耐药,除P1和P2代菌株外,其他代次均出现PAS单一耐药,且耐药水平由低到高依次递增,证明菌株耐药模型诱导成功(表1)。
三、全基因组重测序分析MTB对PAS耐药的遗传进化轨迹
对上述诱导菌株进行全基因组重测序分析,比较耐药菌株与野生型菌株的基因组差异,通过耐药性相关遗传单元特异性分析、突变热点区分析、补偿性突变分析获得与耐药相关的基因组序列位点,如基因内突变、基因间突变等,筛选与耐药相关的基因;通过对所有代次菌株测序,发现PAS耐药可能与3个位点突变直接相关,分别是plcC(Q462R)、folC(S150R)和1个thyA上游位点(3074495G→A)。需要注意的是,诱导过程中,在第三代MTB菌株(P3)获得PAS耐药(MIC为4 mg/L)时,plcC和tlyA上游基因发生突变。至第六代时, MTB菌株(P6)对PAS的MIC上升到8 mg/L,plcC、folC和tlyA上游基因均发生突变。之后至P26,基因组均未在此基础上发生其他突变,但是耐药水平明显提高(表2)。
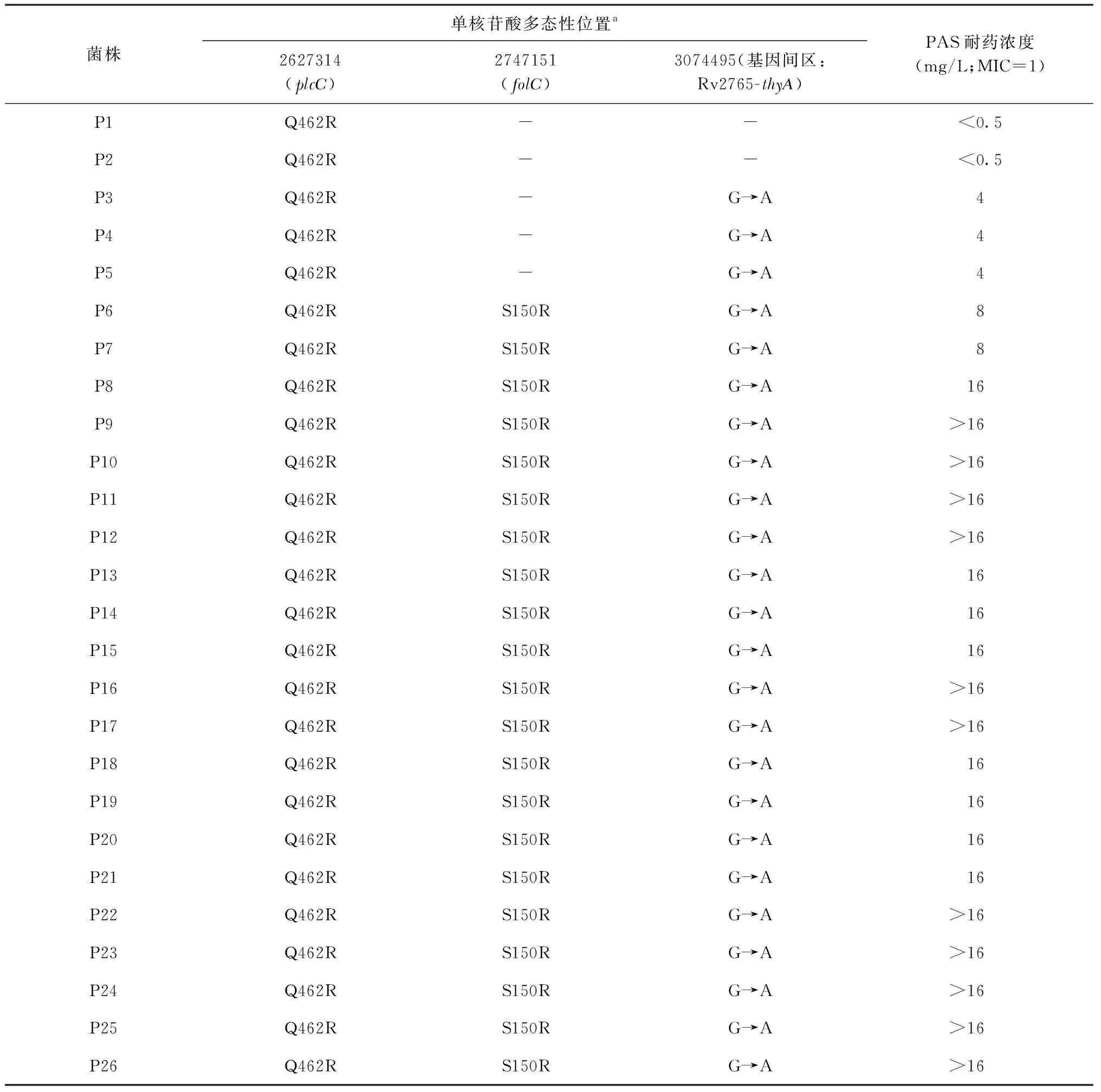
表2 对实验室诱导结核分枝杆菌菌株进行全基因组测序所得的与PAS耐药相关的基因突变
四、全基因组测序分析耐PAS的MTB临床分离株的基因突变
为进一步分析基因突变与MTB耐PAS的关系,筛选耐药水平相似的PAS耐药临床分离株和PAS敏感的临床分离株,并用液体微孔板法进行药敏试验复核。将所筛选到的MTB菌株分为3类:(1)PAS敏感菌株(617株);(2)INH和RFP均敏感、PAS耐药的MTB菌株(30株);(3)INH、RFP和PAS均耐药菌株(72株)。根据流行病学调查数据,发现以上菌株的患者均未使用过PAS进行抗结核治疗。对该719株临床MTB菌株进行全基因组测序,并从突变分析中生成的Bam文件提取各耐药位点的序列比对信息,分析各耐药位点的突变,结合耐药位点数据库中的信息,分析突变和耐药的关系。在617株PAS敏感的MTB菌株中,只发现1株folC基因发生突变(E40G),6株thyA基因发生突变(H75N)。在30株INH和RFP均敏感、PAS耐药的MTB菌株中,folC和thyA均未发生突变。在72株INH、RFP和PAS均耐药的菌株中,3株folC基因突变,3株thyA突变,2株thyX基因间突变(c.-16 C→T)。鉴于菌株来源的患者并未接受过PAS治疗,表明这些临床株中的folC、thyA和thyX突变并非由PAS引起。
讨 论
MTB耐药机制研究虽然是一个老课题,但是,目前的研究几乎都是利用临床分离的敏感菌株和耐药菌株进行研究。由于菌株的遗传背景不同,各菌株的进化水平有差异,因此,难以获知MTB在体外耐药变化的系统进化过程,进而无法全面、准确地分析其耐药机制。本研究中,为了克服背景基因差异的干扰,笔者采用体外药物浓度梯度诱导法将MTB标准菌株H37Rv逐步诱导成不同耐药水平的耐PAS菌株,建立起PAS耐药菌株模型。所有的耐药菌株均筛选自同一标准菌株,基于相同的遗传背景,耐药菌株新积累的遗传变异很可能与其获得的耐药性相关,大大降低了假阳性突变。而且,与高浓度药物诱导的方法不同,该方法可以更好地模拟药物在体内的积累。
全基因组测序技术已经被广泛用于抗结核药物作用机制的研究中[13-16]。通过对本研究中的实验室诱导产生的耐PAS的MTB菌株进行基因组测序,发现由PAS单一因素影响产生的基因突变非常少,包括plcC(Q462R)、folC(S150R)和1个thyA上游位点(3074495G→A),其中2个在PAS耐药相关基因中已有报道。有研究者采用高浓度PAS诱导的方法,也产生了许多folC突变的耐药MTB,其中,Ser150的突变也被包括在内[6, 17]。已知folC可通过形成二氢叶酸(H2Pte-Glu)类似物羟基二氢叶酸(H2PtePAS-Glu)来活化PAS。晶体结构显示,folC突变(S150R)所在的四螺旋束(α1-α2/α4-α5)在与羟基二氢蝶酸(H2PtePAS;H2Pte的类似物)的相互作用中起重要作用,表明该突变体可能影响H2PtePAS谷氨酰胺化的效率。Zhang等[7]研究发现,在分离的临床耐PAS的MTB菌株中存在34.8%(72/208)的folC突变,并且5个特定残基(第40、43、49、150和153位)占临床分离株中folC突变体的94.4%(68/72)。本研究在临床耐PAS的MTB菌株中也观察到几种不同的folC突变(E40G和I43T),但是并未检测到S150R突变。另外,在617株PAS敏感的MTB菌株中,也发现1株folC基因发生突变(E40G),6株thyA基因发生突变(H75N)。而在30株INH和RFP均敏感、PAS耐药的MTB菌株中,folC和thyA均未发生突变。72株INH、RFP和PAS均耐药的菌株中,3株folC基因突变,3株thyA突变,2株thyX基因间突变(c.-16C>T)。需要注意的是,本研究中临床菌株来源的患者从未接受过PAS抗结核治疗,表明这些临床株中的folC、thyA和thyX突变并非由PAS引起。已有研究显示,PAS耐药与大多数抗结核药物(Sm、INH、RFP、EMB、Lfx和Am)的耐药及结核病的治疗史和临床类型(耐多药或者广泛耐药)明显相关[5, 18]。
还有一点值得思考,在实验室诱导的耐PAS菌株中,plcC和tlyA上游基因发生突变后菌株获得低水平PAS耐药(MIC为4 mg/L);plcC、folC和tlyA上游基因均发生突变后,菌株耐药水平升至8 mg/L;之后至P26,基因组均未在此基础上发生其他突变,但是耐药水平明显提高,提示thyA上游位点与PAS低水平耐药相关;累积的folC突变与PAS中度耐药相关;除基因突变外,可能存在其他与对PAS高水平耐药发生相关的调控机制。这具有一定的临床意义,说明可通过优化药物剂量[19]等方法来预防和控制更高水平的PAS耐药发生。
综上所述,本研究通过体外药物浓度梯度诱导法构建了动态连续的PAS耐药MTB菌株模型,并通过全基因组测序发现3个与PAS耐药直接相关的突变位点,间接证明除基因突变因素以外,尚存在其他与PAS高水平耐药发生相关的调控机制,值得进一步研究。对PAS敏感和耐药的MTB临床分离株测序发现,某些folC、thyA和thyX突变并非由PAS引起。另外,对全基因组测序所得的与PAS耐药相关的基因突变也需要进一步验证。
利益冲突所有作者均声明不存在利益冲突
作者贡献余美玲:酝酿和设计实验、实施研究、采集数据、分析/解释数据、文章撰写;张晨晨:酝酿和设计实验、实施研究、采集数据、分析/解释数据;魏文静、赵雨川和卓文基:酝酿和设计实验、文章撰写;郑磊:酝酿和设计实验、文章撰写、工作支持(指导)
