基于加权基因共表达网络鉴定肾移植术后排斥反应中巨噬细胞M1亚型相关基因
2023-02-03董博清李杨石玉婷张静冯新顺郑瑾李潇丁小明薛武军
董博清 李杨 石玉婷 张静 冯新顺 郑瑾 李潇 丁小明 薛武军
与透析相比较,肾移植能明显改善终末期肾病患者的生存质量,并且减少远期并发症[1]。随着肾移植术后新型免疫抑制药的应用,1年移植肾存活率达95%,然而移植肾长期存活率依旧没有明显改善[2]。因此需要明确影响移植物长期存活的危险因素,对具有高危危险因素受者进行早期干预以改善远期结局。
目前,排斥反应仍然是影响移植肾存活的最主要原因[3]。在排斥反应期间,多种细胞浸润移植肾,目前的研究主要集中在适应性免疫细胞,然而,越来越多的证据表明固有免疫中的巨噬细胞与移植肾功能和临床预后密切相关[4-5]。巨噬细胞属于单核吞噬细胞系统,是具有高度可塑性的细胞[6]。经典的巨噬细胞M1亚型,由干扰素(interferon,IFN)-γ,脂多糖和肿瘤坏死因子(tumor necrosis factor,TNF)-α诱导,属于巨噬细胞亚群中促炎细胞亚群,已被证明与移植物预后不良密切相关[7],然而由于巨噬细胞极化的时间以及空间特性,目前对于调控M1亚型极化及募集在移植肾中的具体机制尚不清楚。因此,有必要鉴定排斥反应中参与调控巨噬细胞M1亚型的基因,更加深入了解这些基因如何参与移植肾损伤并影响预后,开发诊断及预后的生物标志物。本研究通过加权基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)以及差异表达分析确定了肾移植术后排斥反应中M1亚型相关的基因,并基于这些基因构建了预测移植肾存活的风险模型。
1 资料与方法
1.1 数据集处理
从基因表达综合(Gene Expression Omnibus,GEO)数据库下载肾移植术后的微阵列数据集GSE36059以及GSE21374[8-9]。两个数据集均基于GPL570平台,GSE36059数据集中包含排斥反应和稳定移植物样本,使用该数据集筛选巨噬细胞M1亚型相关的基因;GSE21374数据集中包含了移植物丢失的随访数据,使用该数据集构建风险模型并进行验证。使用R语言limma包校正两个数据集[10]。
1.2 免疫浸润及加权基因共表达网络分析
CIBERSORT是一种使用基因表达数据估计混合细胞群中成员细胞类型的丰度的反卷积算法。对GSE36059数据集进行CIBERSORT反卷积分析,可计算出各个样本中免疫细胞的含量。WGCNA是用来描述基因关联模式的系统生物学方法,可以用来鉴定高度协同变化的基因集,并根据基因集的内连性和基因集与表型之间的关联鉴定候补生物标记基因。为了筛选出具有共表达性质的基因,使用GSE36059数据集中均绝对误差(mean absolute deviation,MAD)前5 000的基因构建共表达网络。首先进行数据质控,剔除离群样本。然后根据无标度拓扑拟合指数R2以及平均连通性确定最佳的软阈值。通过动态切割树算法识别模块,设置模块内最小基因数为50,将构建的模块聚类以合并相似模块。最后进行相关性分析,分析模块和免疫细胞的相关性,选择和M1亚型相关系数r最高的模块进行后续分析。在模块中选择和模块的|r|>0.8且和M1亚型的|r|>0.4的基因作为M1亚型相关基因。
1.3 差异表达分析
在GSE36059数据集中使用limma包进行差异表达分析,筛选出排斥反应样本和稳定移植物中差异表达基因(differentially expressed gene,DEG)。差异倍数|(Log2 fold change,Log2FC)|>1且校正后P<0.05的基因为DEG。
1.4 基于差异表达的M1亚型相关基因的预后模型构建
将WGCNA以及差异表达分析得到的基因取交集,最终筛选出排斥反应样本和对照样本中差异表达的M1-DEG。随后,按照7∶3拆分GSE21374数据集为训练队列以及验证队列。在训练队列中,使用M1-DEG进行单因素Cox分析,筛选P<0.05的基因构建模型。最小绝对收缩和选择算法(least absolute shrinkage and selection operator,LASSO),是通过生成一个惩罚函数以压缩回归模型中变量的系数,解决严重共线性的问题[11]。将上一步筛选出的基因带入LASSOCox回归中,根据最小均方误差(minimum mean square error,MMSE)选择惩罚系数λ进一步筛选构建风险模型的变量,最后生成列线图可视化模型。
1.5 模型评估

随后通过受试者工作特征曲线下面积(area under curve,AUC)评估基于M1-DEG的风险模型预测1年及3年移植物存活的能力。根据风险评分的中位数将患者分为高风险组和低风险组,使用对数秩检验(log-rank)明确不同组移植物存活是否存在差异。
1.6 免疫浸润及人类白细胞抗原相关基因分析
使用CIBERSORT分析高风险组和低风险组22种浸润的免疫细胞,使用Wilcoxn秩和检验分析组间差异,P<0.05为差异有统计学意义。使用热图描述高风险和低风险组人类白细胞抗原(human leukocyte antigen,HLA)相关基因的分布。
1.7 基因集富集分析
通过基因集富集分析(gene set enrichment analysis,GSEA),进一步阐明高风险组富集的生物学过程以及京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)通路。使用enrichplot包可视化GSEA结果展示标准化富集分数(normalized enrichment score,NES)前5的生物学过程及KEGG通路,P<0.05为差异有统计学意义。
1.8 预测调控预后基因的miRNA
“multiMiR”包集成了7个在线数据库的微小核糖核酸(microRNA,miRNA,miR)和预后基因之间相互作用的信息[12]。为了确保筛选出可靠的和预后基因互作的miRNA,仅纳入出现在≥2个数据库且通过实验证实的相互作用关系。最后,使用可视化软件Cytoscape绘制miRNA-预后基因间的调控关系。
1.9 统计学方法
所有统计分析基于R语言(4.1.2版本)。基因模块与临床特征相关性采用Pearson相关性分析。采用单因素Cox分析筛选与移植物存活密切相关的基因,并采用LASSO-Cox回归分析选择惩罚系数λ进一步筛选构建风险模型的变量。采用log-rank检验分析移植物存活率。采用受试者工作特征曲线评估1、3年移植物存活。双侧P<0.05为差异有统计学意义。
2 结 果
2.1 加权基因共表达网络分析
首先进行数据质控,剔除GSE36059数据集中离群样本(图1A)。根据R2以及平均连通性选择5作为软阈值构建模块(图1B)。通过动态剪切算法最初识别出10个模块,而后合并相似的棕色以及黄色模块(图1C),最终得到9个模块(图1D)。将CIBERSORT计算出的GSE36059各个样本中浸润的免疫细胞的含量和模块进行Pearson相关性分析,其中棕色模块和巨噬细胞M1亚型相关性最高(图1E、F,r=0.45,P<0.001)。最后,筛选出棕色模块中54个和模块及M1亚型相关性高的基因作为M1亚型相关基因(图1G)。

图1 加权基因共表达网络分析Figure 1 Analysis of weighted gene co-expression networks
2.2 确定差异表达的M1亚型相关基因
根据在GSE36059数据集中筛选出42个上调DEG(图2A)。将DEG和WGCNA得到的M1亚型相关基因取交集后,最终得到14个M1-DEG(图2B)。

图2 差异表达的火山图和韦恩图Figure 2 Differentially expressed volcano plots and Wayne diagrams
2.3 基于M1-DEG的风险模型构建
将筛选出的14个M1-DEG带入GSE21374训练集中,使用单因素Cox分析进一步筛选与移植物存活密切相关的基因,14个M1-DEGP<0.05。而后使用LASSO-Cox回归(图3A),根据最小均方误差选择λ(图3B),最终筛选出Toll样受体8(Toll-like receptor 8,TLR8)、Fc γ 受 体 1B(Fc gamma receptor 1B,FCGR1B)、BCL2相关蛋白A1(BCL2 related protein A1,BCL2A1)、组织蛋白酶S(cathepsin S,CTSS)、鸟苷酸结合蛋白2(guanylate binding protein 2,GBP2)及半胱氨酸天冬氨酸蛋白酶招募域家族成员16(caspase recruitment domain family member 16,CARD16)构建风险预后模型[风险 评 分 =−0.921 9×TLR8+(−0.591 7)×FCGR1B+(−1.352 2)×BCL2A1+1.401 7×CTSS+2.056 6×GBP2+0.904 2×CARD16]。使用列线图可视化风险模型预测1年及3年移植物存活,风险评分= −0.921 9×TLR8+(−0.591 7)×FCGR1B+(−1.352 2)×BCL2A1+1.401 7×CTSS+2.056 6×GBP2+0.904 2×CARD16(图3C)。

图3 基于M1-DEG构建风险预后模型Figure 3 Risk prognosis model based on M1-DEG
2.4 模型评估
根据公式计算各个受者的风险评分,根据风险评分中位数将受者分为高风险组和低风险组(图4A、图5A)。在训练集中,风险评分高的受者移植物存活时间短(图4B),基于6个M1-DEG的风险模型预测1年及3年移植物存活的AUC分别为0.918和0.877(图4C)。生存分析表明,高风险组移植物存活率低于低风险组(图4D,P<0.001)。在验证集中的结果和训练集相近(图5B、D),模型预测移植物1年及3年存活的AUC分别为0.765及0.736(图5C)。
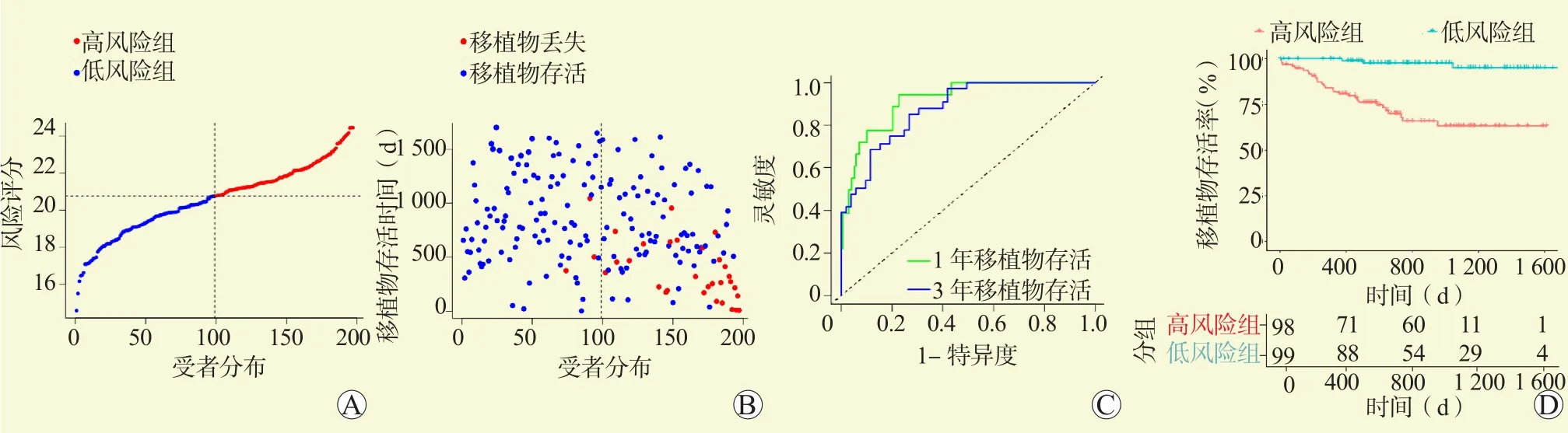
图4 6个基因模型在训练集的表现Figure 4 Six gene models performed in the training set

图5 6个基因模型在验证集的表现Figure 5 Six gene models performed in the validation set
2.5 免疫浸润分析及HLA相关基因分析
在高风险组中,静息及活化的CD4+记忆T细胞、γδT细胞、巨噬细胞M1亚型浸润;而记忆B细胞、巨噬细胞M0亚型、巨噬细胞M2亚型减少(均为P<0.05,图6A)。在高风险组中,HLAⅠ类基因HLA-A、B、C表达上调,部分HLAⅡ类位点的表达上调(图6B)。

图6 高风险组和低风险组的免疫浸润及相关基因分布分析Figure 6 Immune infiltration and related gene distribution analysis between high-risk group and low-risk group
2.6 基因集富集分析
GSEA表明高风险组中明显富集的生物学过程包括免疫反应的活化、适应性免疫反应、基于免疫球蛋白超家族结构域构建的免疫受体体细胞重组的适应性免疫反应、α/β T细胞的活化、α/β T细胞的分化(图7A);高风险组富集的KEGG通路包括移植物排斥反应、抗原提呈和加工、自身免疫性甲状腺疾病、细胞黏附分子、趋化因子信号通路(图7B)。

图7 基因集富集分析Figure 7 Analysis of gene set enrichment
2.7 调控预后基因的miRNA预测
基于7个在线数据库,最后确认了13个miRNA和4个预后基因间的14个相互作用关系,其中CTSS与8个miRNA相互作用、BCL2A1和GBP2与3个miRNA相互作用、FCGR1B与1个miRNA相互作用(图8)。

图8 miRNA和预后基因互作网络Figure 8 miRNA and prognostic gene interaction network
3 讨 论
尽管免疫诱导以及以钙调磷酸酶抑制剂为核心的三联免疫抑制方案提高了移植肾的早期存活率,但移植肾长期存活率仍不理想,排斥反应仍是影响移植肾长期存活的重要因素。巨噬细胞在T细胞介导的排斥反应(T cell-mediated rejection,TCMR)、抗体介导的排斥反应(antibody-mediated rejection,AMR)及慢性排斥反应中均有明显的浸润[13-14],然而目前关于巨噬细胞M1亚型在排斥反应中的作用研究仍然不充分。因此本研究通过生物信息学的方法,分析微阵列数据集,鉴定了排斥反应中和巨噬细胞M1亚型相关的基因,并基于6个M1-DEG构建了预测移植物存活的风险模型。
巨噬细胞具有高度可塑性,其在排斥反应中发挥了多种作用,包括抗原加工和提呈、细胞毒性、吞噬作用及免疫反应的调控等[7]。M1亚型作为促炎细胞的谱系,一方面可以分泌趋化因子以及细胞因子促进炎症[15],另一方面可以促进一氧化氮的产生引起组织损伤,最终影响移植肾的功能[16]。本研究通过WGCNA以及差异分析,在GSE36059数据集初步筛选出14个M1-DEG。随后,在GSE21374的训练集中使用LASSO-Cox回归筛选出TLR8、FCGR1B、BCL2A1、CTSS、GBP2及CARD16构建风险模型。TLR8是Toll样受体家族中的重要成员,与病原体的识别以及先天免疫密切相关,可在肾移植术后诱导无菌性炎症反应并引起T细胞的募集,最终引起排斥反应[17]。一项包括36例肾移植术后急性排斥反应受者的研究,活组织检查结果提示TLR8信使核糖核酸(messenger RNA,mRNA)表达上调[18],与本研究发现一致。TLR8可能通过活化M1亚型以促进其释放炎症因子、产生一氧化氮,从而损伤移植物[19]。
FCGR1B是Fcγ受体Ⅰ家族的成员,其通过结合免疫球蛋白γ的Fc区发挥作用。多中心的前瞻性研究发现肾移植受者血液中的FCGR1B的表达量与AMR的发生相关[20],AMR受者外周血中FCGR1B的mRNA表达显著上调。既往研究发现小鼠的巨噬细胞和树突状细胞高表达FCGR1[21],因此FCGR1一方面可以与免疫球蛋白结合在体液免疫中发挥作用,另一方面可以通过介导非T细胞依赖的免疫复合物的摄取,联系适应性免疫和固有免疫。BCL2A1和细胞凋亡的进程密切相关,在使用HLA预制抗体处理肾微血管内皮细胞的研究中,加入IFN-γ和TNF-α组中BCL2A1的蛋白水平表达升高[22]。此外,在一项应用支气管肺泡灌洗液研究细胞因子风暴的单细胞分析中,BCL2A1和巨噬细胞的炎症反应以及趋化作用相关[23],进一步表明该基因可能通过影响巨噬细胞的募集过程而影响移植物的存活。CTSS编码的蛋白是肽酶C1家族的成员,其参与了抗原蛋白的降解呈递给MHCⅡ类分子。一项小鼠心脏移植研究中使用了CTSS抑制剂,结果发现实验组小鼠移植物存活时间更长,且没有产生供体特异性抗体(donor specific antibody,DSA)[24]。同样,在一项异基因小鼠脾脏移植研究中,移植脾脏组织CTSS表达量显著上升,并且应用CTSS抑制剂减少了T细胞的活化[25]。除了通过抗原提呈活化CD4+T细胞,CTSS也可以调控固有免疫诱导巨噬细胞释放白细胞介素(interleukin,IL)-1β[26]。GBP2与IFN-γ信号传导途径有关,既往研究表明移植肾组织中该基因表达量可以用于诊断肾移植术后排斥反应[27]。在一项包括46例肝移植受者的研究中,发生急性细胞性排斥反应(acute cellular rejection,ACR)的受者外周血白细胞中GBP2 mRNA表达显著上调,且多因素分析也表明GBP2高表达是ACR的独立危险因素[28]。CARD16编码的蛋白与半胱氨酸天冬氨酸蛋白酶(cysteinyl aspartate specific proteinase,Caspase)的激活以及随后的IL-1β释放相关。既往研究认为CARD16是Caspase的抑制剂,抑制了后续的炎症发展[29]。然而也有研究报道寡聚化的CARD16是Caspase的激活剂[30]。因此,CARD16可能通过影响巨噬细胞分泌IL-1β参与到移植物的损伤过程中。基于6个M1-DEG的风险模型对于预测移植肾的存活具有良好的表现,生存分析表明高、低风险组的受者移植肾的存活存在明显差异。因此,这些生物标志物有助于在早期对肾移植受者进行风险分层,确定移植肾丢失高风险受者,从而给予抢先干预,改善远期预后。
在高风险组中,静息及活化的CD4+记忆性T细胞、γδT细胞及巨噬细胞M1亚型浸润增多。巨噬细胞通过将抗原提呈给CD4+T细胞使其活化,活化的CD4+T细胞通过产生IFN-γ和IL-2驱动TCMR,并产生IL-4、IL-5和IL-13参与AMR[31]。γδT细胞是固有免疫的重要组成部分,该细胞和移植后的排斥反应及巨细胞病毒(cytomegalovirus,CMV)感染相关[32]。既往研究发现在肾移植术后排斥反应期间移植肾组织中γδT细胞明显浸润,占总T细胞的10%以上[33]。在肾移植术后CMV感染后,循环中产生的CMV特异性γδT细胞有助于病毒的清除[34]。这些γδT细胞高表达CD16,因此其可能通过抗体依赖性细胞毒性参与由DSA促进的同种异体移植损伤[35]。值得注意的是,在高风险组的移植肾组织中M1亚型浸润增多,而M0以及M2亚型减少,这揭示了巨噬细胞亚群平衡的失调是移植肾损伤的重要机制。GSEA分析进一步揭示了在高风险组中,免疫反应、排斥反应、细胞黏附分子及趋化因子信号通路明显富集。最后,使用数据库预测了调控预后基因的miRNA,这将为后续进一步分析预后基因如何影响M1亚型的极化并造成移植物损伤提供依据。
综上所述,本研究鉴定了排斥反应中巨噬细胞M1亚型相关的基因,并构建了风险模型,对于预测移植肾存活具有良好的表现,为早期鉴定高风险受者提供依据,但仍存在一些局限性。微阵列数据集无法阐明巨噬细胞极化过程中更加详细的时间以及空间特征[6],将巨噬细胞简单地划分为M1和M2两个亚型无法满足生物体内不同组织以及病理状态[36]。供者以及受者来源的巨噬细胞可能在排斥反应中发挥了不同的作用,本研究无法区分巨噬细胞的来源,且需要后续的动物实验进一步验证分子机制。
