Sn调变Cu电子结构改善Cu/CeZrO2抗硫性能
2023-02-02宋天远梁美生李巧艳叶翠平
宋天远,梁美生,李巧艳,杨 超,叶翠平
(太原理工大学 环境科学与工程学院,太原 030024)
近年来,汽车尾气成为大气污染的主要污染源之一[1],随着汽车污染管理规定的日趋严格,三效催化剂(TWC)可以有效去除汽车尾气中的CO、NOx和CH化合物三种主要污染物,并在汽车生产中得到广泛的应用。但是我国汽油类产品中的硫含量高,其燃烧生成的SO2等硫化物将显著影响TWC去除尾气污染物的效率,国内外科研工作者对SO2的毒化作用作了大量的研究,发现SO2会导致催化剂活性下降甚至完全丧失[2-7]。人们普遍认为,下一代催化剂需要具有一定的抗硫性能,以保证催化剂的催化性能[8-9]。目前,减轻三效催化剂硫中毒的方法主要有以下三种:改善油品质量、在催化剂中加入“SO2诱捕剂”以及改变催化剂对SO2的吸附及活化强度[10]。加入诱捕剂的方式是通过优先吸附SO2来保护活性组分,它只能短时间延长催化剂的寿命。改变SO2吸附及活化强度可以抑制硫酸盐沉积到催化剂活性组分颗粒外壳,降低硫中毒几率。三效催化剂是由活性金属、助剂以及载体三部分组成,不同部分之间的协同作用一直是研究的热点,特别是通过助剂改性提高催化剂性能[11],其对性能的提升归因于几何结构变化或电子相互作用[12],这也是最直接、有效的方法[13]。LIU et al[14]将单原子Pd负载到CeO2(110)表面,掺杂Cu进行助剂改性,证明助剂的改性能够降低反应活化能,提高催化剂的CO氧化性能。
传统的汽车尾气催化剂均以贵金属为活性组分,虽然该类催化剂具有较高的活性,但贵金属催化剂存在成本较高、在高温下易烧结失活、抗硫性差的缺点。因此,许多研究人员致力于开发高效且具有抗硫能力的以非贵金属为活性组分的汽车尾气催化剂。Cu基催化剂因其特殊的催化能力在多个催化领域里受到广泛关注,在水煤气变换(WGS)[15]、低温下CO氧化[16-18]及CO选择性催化还原NO[19]等反应中都表现出优良的催化性能。近年来,Cu基催化剂也广泛应用于汽车尾气处理体系,但是研究发现铜基催化剂的抗硫性能差,生成的CuSO4覆盖活性位点导致不可逆硫中毒[20],催化剂的催化性能大幅度下降。CeZrO2由于具有优异的储氧能力通常被用作Cu基三效催化剂的助剂,而且有研究表明,对CeZrO2助剂进行改性可以在一定程度上减轻SO2的吸附强度,提高铜基催化剂的抗硫中毒性能[21]。在助剂中引入第三种元素,是提高催化剂抗硫性能最简便有效的方法[17,22-23]。
相对较低的温度下,SnO2能够在+4和+2价之间快速且可逆地相互转化[24-25],双价有助于表面氧组成的变化,因此也可以用作助剂[26]。HAO et al[10]制备了一系列Sn改性CeMoOx催化剂,与CeMoOx相比,Sn0.2CeMoOx具有更小的晶粒尺寸和更高的表面酸度,减弱了SO2在催化剂上的吸附,显著提高催化剂的抗硫性能。CHANG et al[27]发现与MNOx-CeO2相比,Sn改性的MNOx-CeO2对SO2以及对H2O的抗性显著提高。目前的研究热点集中于CeO2(111)与(110)面,与CeO2(111)相比,CeO2(110)表面具有更高的表面能和较低的氧空位形成能[28-31],而且它通常暴露在催化材料的纳米棒[32]和纳米颗粒[33]上。YANG et al[34]研究了CeO2(110)上的氧空位缺陷,发现它比CeO2(111)更具反应性,与CeO2(110)更容易还原的事实相吻合[35],说明金属负载的CeO2(110)表面将表现出更优异的催化活性。基于此,本论文对较活泼并且稳定的CeZrO2(110)面进行研究,结合密度泛函理论计算系统探讨Sn改性前后Cu/CeZrO2(110)催化剂表面性质和构效关系的变化,进一步揭示对其抗硫性能的影响,为优化设计新型高性能汽车尾气催化剂指明方向。
1 DFT计算方法
所有的计算工作均采用Materials Studio软件包中的Dmol3模块完成[36]。交换相关势采用密度泛函理论中的广义梯度近似和Perdew-Burke-Ernzerhof泛函相结合的方法(GGA-PBE)[37-38]。Cu、Ce、Zr和Sn金属原子的内层电子采用半核芯赝势(DSPP),价电子波函数采用双数值基加极化函数(DNP)展开,O原子采用全电子(All electron)基组[39]。在几何优化计算中,能量、力和位移的收敛标准分别为10-5Ha、0.002 Ha/atom和0.000 5 nm.实空间截断半径保持为0.58 nm,拖尾效应(Smearing)设置为0.005 Ha.
本文首先对Ce0.75Zr0.25O2晶胞进行几何优化,优化得到的晶格参数(a=b=c=0.540 43 nm)与文献中报道的实验值和计算值相吻合[40-41]。对于CeZrO2(110)表面,采用p(2×2)的超晶胞,超晶胞有6个原子层,固定底部三层原子,其余原子和表面吸附物弛豫。为确保模型层与层之间没有相互作用,在z方向上设置1.8 nm的真空层。布里渊区积分Monkhorst-Pack网格参数设为2×3×1.所有的体系都考虑了自旋极化。优化单个SO2分子与Cu原子时,将表面模型设置为1.08 nm×0.76 nm×2.76 nm周期边界模型。SO2的键长为0.148 1 nm,键角为119.843°,与先前的理论计算结果一致[42-43]。
吸附在表面的一个铜原子的吸附能定义如下:
Eads(Cu)=ECu/M-EM-ECu.
(1)
其中EM、ECu和ECu/M分别对应CeZrO2载体、自由态Cu原子、Cu负载到载体的能量。ECu的计算采用与吸附体系相同的原胞。根据公式(1)中Eads(Cu)的定义,Eads(Cu)值越负,吸附作用越强。
考虑到每个SO2吸附在Cu负载的表面上,可以引入类似的公式:
Eads(SO2)=ESO2/M-EM-ESO2.
(2)
其中EM、ESO2和ESO2/M分别代表M衬底、孤立的SO2、衬底吸附SO2的总能。对ESO2的计算采用与吸附体系相同的原胞,可以发现,Eads(SO2)<0表示放热过程,Eads(SO2)>0表示吸热过程。
2 结果与讨论
2.1 Cu在CeZrO2(110)和CeZrSnO2(110)表面的吸附
2.1.1Cu在CeZrO2(110)表面的吸附
为了找出Cu在CeZrO2(110)表面的各种可能吸附模型,将单个Cu原子放置在CeZrO2(110)表面多个位点进行优化,得出六种稳定的吸附模型,图1中显示了六种稳定的吸附模型,分别为O—O(表面O桥位点)、Zr—Zr(表面Zr桥位点)、Ce—Ce(表面Ce桥位点)、Ou(表面O顶点)、Zru(表面Zr顶点)、Ceu(表面Ce顶点)。棕色、红色、淡黄色和蓝色球体分别代表Cu、O、Ce和Zr原子。所有稳定构型都列于图1中,并在表1中同时列出了吸附能与相应的关键几何参数。

图1 Cu在CeZrO2(110)表面的可能吸附构型:Zr—Zr(锆桥),Ou(氧顶),O—O(氧桥),Zru(锆顶)Ceu(铈顶)Ce—Ce(锆桥)Fig.1 Structural models of Cu adsorbed on CeZrO2(110): Zr—Zr(Zr-bridge),Ou(O-top),O—O(O-bridge),Zru(Zr-top)Ceu(Ce-top),Ce—Ce(Ce-bridge)

表1 Cu吸附在CeZrO2(110)上的吸附能、键长Table 1 Adsorption energy and bond length of Cu adsorbed on CeZrO2(110)
从表1可以看出,所有吸附能均为负值,表明Cu在CeZrO2(110)表面的吸附是放热过程。Cu的最稳定的吸附位点为O—O,即氧桥位且是次层Ce的顶位(记为O—O位),它具有-3.52 eV的最大吸附能。最稳定结构中两个Cu—O键键长均为0.178 nm,与实验上Cu2O晶格中Cu—O的键长(0.186 nm)相接近[44]。次稳定吸附位点在锆桥(Zr—Zr)位,吸附能为-3.41 eV,比最稳定的吸附位点对应的吸附能小0.11 eV.在Ce—Ce、Ou、Zru和Ceu位点,Cu的吸附能分别为-2.44 eV、-1.59 eV、-0.43 eV和-0.25 eV,Cu原子与表面M原子的Cu—M键长度分别为0.211 nm、0.195 nm、0.279 nm和0.311 nm,Cu原子与CeZrO2(110)表面有较远距离,其吸附能较小。
为了进一步研究吸附过程中Cu原子与CeZrO2(110)表面之间的相互作用,本论文基于Cu的最稳定吸附构型(O—O),对被吸附前后Cu的分波态密度(PDOS)进行分析,结果如图2所示。从图2可以发现,被吸附前,Cu原子的4 s轨道在费米能级显示一个峰,而Cu原子3 d轨道在-0.27 eV处被占据。吸附Cu后,Cu原子的s、d轨道强度变低,峰变宽,并且所有轨道都将移动到较低的能级。综上所述,Cu原子与Cu/CeZrO2(110)之间有很强的相互作用[18]。
2.1.2Cu在CeZrSnO2(110)表面的吸附
之后将第一层的一个Ce原子用Sn原子代替,进行几何优化后,将Cu原子吸附到CeZrSnO2(110)表面进行优化,图3为CeZrO2(110)、Cu/CeZrO2(110)、CeZrSnO2(110)、Cu/CeZrSnO2(110)的吸附模型。棕色、红色、淡黄色、蓝色和褐色球体分别代表Cu、O、Ce、Zr和Sn原子。表2为CeZrO2(110)、Cu/CeZrO2(110)、Cu/CeZrSnO2(110)、CeZrSnO2(110)中部分原子的Mulliken电荷数。

图2 分波态密度Fig.2 Partial density of states (PDOS)
由图3可知,在Cu/CeZrO2和Cu/CeZrSnO2体系中,Ce—O之间的键长范围为0.336~0.430 nm,远大于在立方CeO2中Ce4+—O的理论平均键长(0.235 nm)(晶格参数为0.542 nm)[45]和实验平均键长(0.234 nm)(晶格参数为0.541 nm)[23],这表明在吸附过程中Ce(编号1)发生了Ce4+还原为Ce3+反应。同时,根据表2发现Cu吸附前后各个原子的电荷转移情况:CeZrO2体系在Cu吸附前后,氧桥位下的次层Ce原子得到0.155个电子,被还原为Ce3+,这些电子来自于吸附的Cu、第一层的两个Ce和两个Zr;而CeZrSnO2体系在Cu吸附前后,由于Sn离子半径小,比Ce难失去电子,当第二层Ce被还原时,Sn不能像Ce一样提供那么多电子,这些电子由吸附的Cu代为提供,形成了在Cu/CeZrSnO2体系中活性组分Cu具有更强正电性的现象。根据之前的DFT计算,SO2优先与正电性更弱的Cu结合,而Sn的掺杂增强了Cu的正电性,减弱了SO2吸附能力,进而提高了催化剂的抗硫性能[46]。

图3 CeZrO2(110)、Cu/CeZrO2(110)、CeZrSnO2(110)、Cu/CeZrSnO2(110)的模型Fig.3 Structural models of CeZrO2(110),Cu/CeZrO2(110),Cu/CeZrSnO2(110),CeZrSnO2(110)
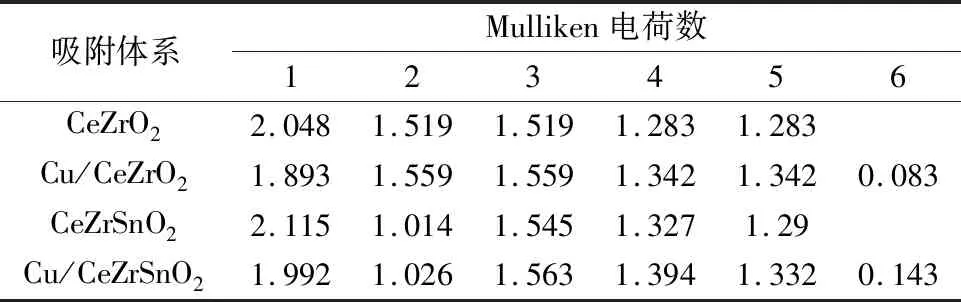
表2 CeZrO2(110)、Cu/CeZrO2(110)、CeZrSnO2(110)、Cu/CeZrSnO2(110)中部分原子(用数字1-6进行标记)的Mulliken电荷数Table 2 Mulliken charge of some atoms (marked with numbers 1-6) in CeZrO2(110),Cu/CeZrO2(110),Cu/CeZrSnO2(110),CeZrSnO2(110)
图4及图5为总态密度及分波态密度图。从图4的CeZrO2(110)、CeZrSnO2(110)面的总态密度(TDOS)表明,CeZrO2(110)带隙约为1.24 eV,CeZrSnO2(110)带隙约为1.06 eV.与之相比,Cu吸附CeZrO2(110)表面的带隙为1.00 eV,带隙变窄(0.24 eV),Cu吸附的CeZrSnO2(110)表面的带隙为0.90 eV,带隙变窄(0.16 eV)并出现了一些间隙态(见图4).通常这些间隙态的出现是由外来金属的吸附引入的[18],因此称为MIGS(metal-induced gap states),本论文是由于Cu的引入出现的这种现象。MIGS位于价带与费米能级之间,结合图5分波态密度分析发现:MIGS主要来自于Cu3d态及与其邻近的O2p态和表层一个Ce的4f态、Zr离子的3d态。
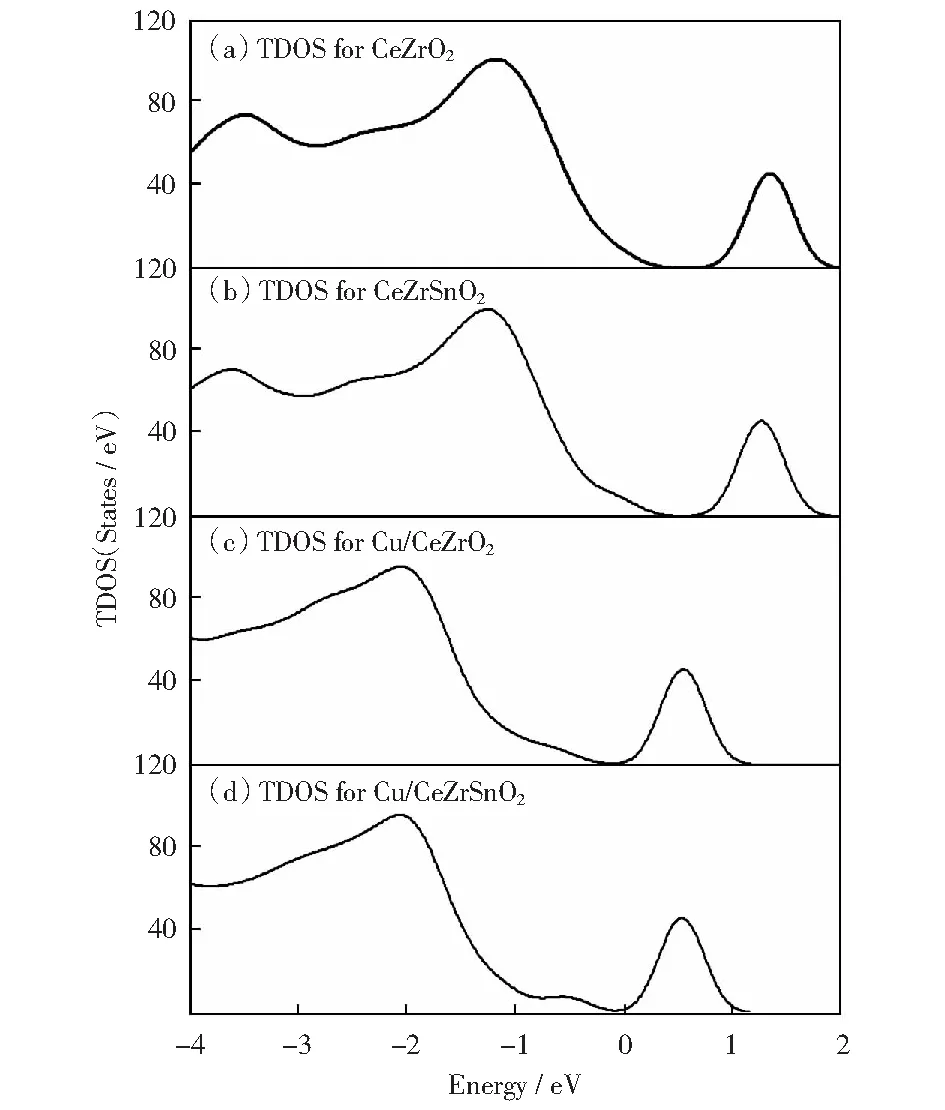
图4 总态密度Fig.4 TDOS
图5给出了CeZrO2、CeZrSnO2、Cu/CeZrO2以及Cu/CeZrSnO2的Cu和其表面的Ce、O、Zr的分波态密度(PDOS).从图5(a)、(b)我们发现,Sn的3d态与其近邻CeZrO2(110)面之间存在电子杂化,说明Sn成功掺杂进CeZrO2(110)的内部,这主要是因为掺杂的Sn4+与CeZrO2体系中的Ce4+、Zr4+有相同的价态,容易将Ce替换为Sn[47].从图5(c)、(d)可以看出,吸附了Cu之后,Cu的DOS的两个主峰都靠近费米能级,说明Cu的PDOS的局域性很强。比较Cu及其近邻O的分波态密度图发现,Cu的3d态与其近邻O的2p态之间出现电子杂化,这也是Cu与衬底强烈相互作用的根源,掺杂Sn后杂化现象更加强烈,这也是Cu/CeZrSnO2的MIGS更加明显的原因。基于以往关于Cu—CeO2、Pd—Ce0.75Zr0.25O2、Pt—Ce0.75Zr0.25O2[18,48]体系的结果,MIGS对催化剂的催化性能很重要,它能够提高储氧能力,使氧化还原反应更容易发生。这说明Sn的掺杂增强了Cu与O的相互作用,可能提高催化剂催化性能。此外,可以看出,掺杂Sn(图5d)后的Cu—3d轨道靠近费米能级处的峰值下降,能带宽度变窄,离域性减弱,表明Cu与周围原子的成键减弱,与之后对SO2的吸附减弱一致。

图5 分波态密度Fig.5 PDOS
2.2 SO2在Cu/CeZrO2(110)表面和Cu/CeZrSnO2(110)表面的吸附行为
在汽车尾气中,SO2的存在会造成汽车尾气催化剂不可逆的硫中毒。本节集中研究SO2在Cu/CeZrO2(110)表面和Cu/CeZrSnO2(110)表面上的吸附,如图6所示。其中,棕色、红色、淡黄色、蓝色、灰色和深黄色球体分别代表Cu、O、Ce、Zr、Sn和S原子。SO2的吸附能、夹角、键长以及S所带电荷列于表3中,其中吸附能(Eads(SO2))由公式(2)得出。

图6 SO2吸附在Cu/CeZrO2(110)和Cu/CeZrSnO2(110)的模型图Fig.6 Structural models of SO2/Cu/CeZrO2(110) and SO2/Cu/CeZrSnO2(110)

表3 SO2吸附在Cu/CeZrO2(110)和Cu/CeZrSnO2(110)上的吸附能、键角、键长、S所带电荷和Cu电荷变化Table 3 Adsorption energy, bond angle, bond length, S charge and Cu charge change on Cu/CeZrO2(110) and Cu/CeZrSnO2(110)
表3表明,与Cu/CeZrO2(110)相比,Cu/CeZrSnO2(110)对SO2的吸附较弱,吸附能为-0.85 eV[49]。将Sn引入Cu/CeZrO2催化剂后,SO2中毒的能力减弱。如表3所示,与游离SO2相比,SO2吸附在Cu/CeZrO2(110)或Cu/CeZrSnO2(110)上后,S—O的键长(dS—O)增加,O—S—O角减小。在Cu/CeZrO2(110)上,S—O的键长(dS—O)增加0.000 8 nm,O—S—O角减小0.05°.在Cu/CeZrSnO2(110)中,S—O长度增加0.045 0 nm,O—S—O角减小3.77°.这些几何变化可能是由于SO2活化引起,活化可能会引发S—O键的解离和金属催化剂上中间体的形成[43]。
表3显示了吸附在Cu/CeZrO2(110)和Cu/CeZrSnO2(110)上的SO2的净电荷以及Cu电荷的变化。与SO2/Cu/CeZrO2(110)体系相比,SO2/Cu/CeZrSnO2(110)体系SO2中S的电荷为0.516,与SO2/Cu/CeZrO2(110)体系SO2中S原子所带电荷相比,失去较少电子,Cu仅得到了0.007个电子,比CeZrO2(110)体系中Cu原子的电荷变化小(0.024).可见,当SO2吸附在Cu/CeZrO2(110)上时,某些来自S原子的电子转移到Cu原子上,但仍然是SO2/Cu/CeZrSnO2(110)体系中Cu原子显示更强的正电性,不利于Cu和S原子之间的电子转移,从而提高了Cu的抗硫性能[49]。
为了进一步了解Sn在Cu—Sn系统中的作用,本论文采用净电荷,分波态密度(PDOS)和d带中心理论分析电子结构信息,结果如图7所示。与图7(b)相比,SO2上的S-p PDOS的峰值比Cu—Sn(图7(d))的S-p PDOS移向更负能量的地方,这表明SO2和Cu—Sn之间的相互作用较弱。吸附SO2后,Cu中Cu—d的靠近费米能级处的峰由-0.68 eV后移到-1.33 eV并且峰值下降很明显,而在Cu—Sn中该峰仅仅从-0.52 eV移动到-0.88 eV,峰值几乎没有改变。不管SO2如何吸附,Cu(Sn)—d几乎都保持其原始PDOS轮廓,这表明SO2和Cu/CeZrSnO2(110)之间的相互作用较弱。

图7 分波态密度Fig.7 PDOS
根据HAMMER和NØRSKOV[50-51]的化学吸附模型,d带中心与分子表面吸附强弱密切相关。表4显示了SO2在Cu/CeZrO2(110)和Cu/CeZrSnO2(110)上吸附之前和之后d带中心的变化。在一定程度上,d带中心显示了金属的d轨道能级,它是确定催化剂与吸附质结合能力和预测催化剂吸附能力的重要参数。d带中心和吸附能的关系主要是通过d带中心靠近还是远离费米能级来判断,通常来说,d带中心越高越靠近费米能级,吸附更稳定。由表4可知,Sn的加入会使得d带中心远离费米能级,这表明Sn的引入会削弱SO2和Cu之间的相互作用。

表4 Cu/CeZrO2与Cu/CeZrSnO2表面原子的d带中心Table 4 The d-band center of Cu/CeZrO2 and Cu/CeZrSnO2 surface atoms eV
3 结论
本文通过DFT理论计算Sn掺杂Cu/CeZrO2(110)催化剂的电子结构和抗硫性能之间的关系。DFT计算结果表明,Cu在CeZrO2(110)的氧桥处吸附为最稳定的吸附模式,吸附能为-3.52 eV.由MIGS可知,Sn掺杂后的Cu3d态及与其邻近的O2p态重叠更多,表明Sn掺杂可以增强Cu与O之间的相互作用。与Cu/CeZrO2(110)相比,在Cu/CeZrSnO2中,Cu表现出更强的正电性,同时通过对SO2在Cu/CeZrO2(110)和Cu/CeZrSnO2(110)上的电荷以及PDOS比较,认为Cu的正电性阻止电子从S到Cu的迁移,减弱SO2的吸附强度,进而提高催化剂的抗硫性能。
致谢:本论文的模拟计算由上海超算科技有限公司协助完成,在此表示感谢。
