去屑洗发露中羟吡啶酮的高效液相色谱法检测及质谱确证
2023-01-24聂明霞梁文耀夏泽敏陈彦君谭建华
聂明霞,汪 毅,梁文耀,贾 芳,李 露,夏泽敏,陈彦君,谭建华
(广州质量监督检测研究院,国家化妆品质量检验检测中心,广东 广州 511447)
羟吡啶酮为1-羟基-2-吡啶酮类化合物,因含有与曲霉酸相似的异羟肟酸结构而具有抑菌作用[1]。有报道称该类化合物可用来治疗溢脂性皮炎[2]、口腔黏膜疾病[3]及由真菌、细菌引起的皮肤感染疾病[4],其作为配位体与2-氨乙基等化合物合成的铁离子螯合剂也具有抑菌效果[5]。然而,相关毒理学试验表明该化合物具有潜在的致突变性[6-7]。因此,《化妆品安全技术规范》(2015年版)[8]中明确规定,禁止在化妆品中使用羟吡啶酮。但目前尚无化妆品中羟吡啶酮的测定方法报道。
羟吡啶酮的极性强,反相色谱保留能力较弱,且和2-羟基吡啶-N-氧化物(HOPO)存在互变异构现象[9-10](图1)。而HOPO与色谱系统中的金属离子可能发生较强的络合作用,两种互变异构形态在色谱柱上的异构互变会引起色谱峰严重拖尾或分峰现象,从而给色谱分析带来困难。本文以去屑洗发露为考察基质,通过优化仪器方法及提取条件等,解决了羟吡啶酮难以保留、峰形差和杂质干扰等问题。同时,采用化妆品常用的液相色谱-串联质谱检测技术[11],结合硫酸二甲酯衍生化处理,建立了一种洗发露中禁用原料羟吡啶酮的快速、准确、灵敏的测定方法和质谱确证方法,以为化妆品的安全监管提供有效的技术支撑。
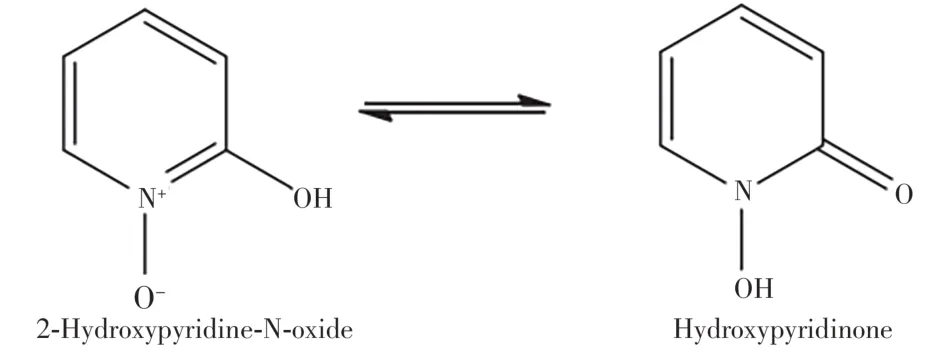
图1 HOPO两种形态的互变异构Fig.1 Tautomerization of HOPO
1 实验部分
1.1 仪器与试剂
Agilent 1260高效液相色谱仪,配DAD检测器(美国安捷伦公司);岛津液相色谱仪-SCIEX AB 5500+三重四极杆质谱仪(日本岛津与美国SCIEX公司);BSA224S-CW电子天平(德国赛多利斯公司);MS3 basic涡旋振荡器(德国IKA公司);KQ-250DV型数控超声波清洗仪(昆山市超声仪器有限公司);Milli-Q纯水系统(美国Millipore公司)。
羟吡啶酮(纯度为95.0%,CAS:822-89-8,美国IL及Macklin公司);2-羟基吡啶-N-氧化物(纯度为98.0%,CAS:13161-30-3,美国Aladdin及Mackin公司);甲醇、乙腈(色谱纯,德国Merck公司);草酸、三乙胺(分析纯,广州化学试剂厂);硫酸二甲酯(分析纯,山东西亚化学工业有限公司);实验用纯水(18.2 MΩ·cm)由Milli-Q纯水系统制备。
1.2 标准溶液的配制
精密称取羟吡啶酮对照品约0.1 g(精确至0.1 mg),置于50 mL烧杯中,加适量乙腈溶解后转移至100 mL容量瓶中,用乙腈稀释至刻度,混匀,即得质量浓度为1 000 mg∕L的羟吡啶酮标准储备液。
分别精密移取不同体积的标准储备液于10 mL容量瓶中,用乙腈定容,配制成质量浓度分别为1.0、2.0、5.0、10.0、50.0、100.0 mg∕L和10.0、20.0、50.0、100.0、200.0、500.0 µg∕L的标准曲线工作溶液。
1.3 样品前处理
称取样品0.5 g(精确至0.001 g)于10 mL具塞比色管中,加入1 mL乙腈,涡旋30 s使样品分散,再用乙腈定容至刻度,涡旋30 s后,超声提取15 min。冷却至室温后,上清液过0.22 µm滤膜,待测定。
1.4 仪器分析条件
色谱柱:Waters Atlantis Hilic(4.6 mm × 250 mm × 5 µm);流动相:950 mL乙腈 + 50 mL 400 mmol∕L草酸溶液;流速:1.0 mL∕min;进样量:20 µL;柱温:30 ℃,检测波长:306 nm。
1.5 液相色谱-串联质谱确证方法
1.5.1 标准曲线衍生制备移取“1.2”中10.0、20.0、50.0、100.0、200.0、500.0 µg∕L的羟吡啶酮标准溶液1 mL于比色管内,加入0.5 mL 0.3 mol∕L NaOH 溶液,涡旋30 s后,加入50 µL硫酸二甲酯,涡旋30 s,于37 ℃水浴中加热15 min后,再加入50 µL三乙胺,涡旋30 s,取衍生后的标准溶液用纯水稀释10倍,过0.22 µm滤膜,待测定。
1.5.2 样品前处理与衍生方法取“1.3”中的样液1 mL于比色管中,加入0.5 mL 0.3 mol∕L NaOH溶液,涡旋30 s后,加入50 µL硫酸二甲酯,涡旋30 s,于37 ℃水浴中加热15 min后,再加入50 µL三乙胺,涡旋30 s,取样液用纯水稀释10倍,过0.22 µm滤膜,待测定。
1.5.3 液相色谱条件色谱柱:Agilent SB C18(2.1 mm × 100 mm,2.7 µm);流动相:A为0.1%甲酸水溶液,B为乙腈,等度洗脱,VA∶VB= 9∶1;流速:0.3 mL∕min;进样量:2.0 µL;柱温:30 ℃。
1.5.4 质谱条件离子源为电喷雾离子源(ESI源);监测模式为正离子多反应监测(MRM);离子化电压4 500 V;气帘气137.8 kPa;喷雾气344.7 kPa;碰撞气62.1 kPa;离子源温度450 ℃;监测离子对、碰撞电压(CE)和去簇电压(DP)参数见表1。

表1 羟吡啶酮的质谱参数Table 1 Mass spectrometric parameters of hydroxypyridone
2 结果与讨论
2.1 仪器方法的建立
2.1.1 色谱柱的选择羟吡啶酮的logP值为-0.65,亲水性强,极性较大,难以在色谱柱上获得较好保留,因此对比了几种规格相同(4.6 mm × 250 mm × 5 µm)的C18柱(Pronto SIL-C18、Ultimate AQ C18)和Waters Atlantis Hilic色谱柱的效果。以100%水为流动相对2 µg∕mL羟吡啶酮标准溶液进行分析发现,C18柱对羟吡啶酮的保留较弱,且色谱峰拖尾难以消除,因此难以对低浓度羟吡啶酮准确定量;而使用Hilic色谱柱时目标物的保留时间较使用C18柱有所延长,峰形更加对称,因此选用Hilic柱进行后续方法开发。
2.1.2 检测波长的选择采用配备二极管阵列检测器的高效液相色谱仪对标准溶液进行检测,羟吡啶酮的紫外吸收光谱图显示,其特征吸收波长为228 nm和305 nm,与用乙醇溶解的羟吡啶酮及其互变异构体HOPO的特征吸收波长一致[1]。在实际样品测试过程中,228 nm波长下的基质干扰较大,而羟吡啶酮在305 nm波长处的响应值满足方法灵敏度与稳定性要求,因此选择305 nm作为检测波长。
2.1.3 流动相的选择比较了乙腈-水、乙腈-0.1%甲酸水溶液、乙腈-20 mmol∕L醋酸铵水溶液、乙腈(含0.1%甲酸)-甲醇和乙腈-草酸水溶液5种流动相体系对羟吡啶酮的分离效果。结果显示,使用前3种流动相对羟吡啶酮进行分析,色谱峰均存在不同程度分峰现象;使用95%乙腈(含0.1%甲酸)-5%甲醇作流动相时,分峰现象有所改善,但拖尾仍然严重;采用乙腈-10 mmol∕L草酸水溶液为流动相,羟吡啶酮的分峰和拖尾现象被有效消除,色谱峰形良好。这可能是由于草酸水溶液能够大大降低色谱柱上残留金属离子与化合物的络合作用所致。
进一步比较了不同浓度的草酸(5、10、20、40 mmol∕L)对羟吡啶酮的色谱分离效果。结果显示,随着草酸浓度增大,羟吡啶酮的保留时间有所增加,草酸浓度达20 mmol∕L后,保留时间无明显延长。考虑到高浓度草酸溶液在高比例乙腈中有析出风险,最终选择950 mL乙腈 + 50 mL 400 mmol∕L草酸溶液为流动相,预先混合后使用。
2.1.4 液相色谱-串联质谱确证方法由于洗发水的配方组分复杂,常用的吡硫鎓锌、吡罗克酮等去屑剂与羟吡啶酮有相似的特征吸收,仅依靠保留时间和紫外吸收光谱定性可能会因存在干扰而产生假阳性。因此,建立了羟吡啶酮的液相色谱-串联质谱确证方法,对阳性样品或疑似阳性样品进行确证,以保证检测结果的准确性。
直接使用液相色谱-串联质谱对羟吡啶酮标准样品进行分析时,其质谱响应极弱,过高的检出限无法满足分析要求。将硫酸二甲酯在碱性条件下与氨基羟基发生反应[7],使化合物甲基化,可提高电离效率,从而大大增强化合物的质谱响应,且解决了化合物的分峰拖尾问题,满足分析确证要求。确证条件见“1.5”。
2.2 前处理方法的优化
2.2.1 提取溶剂的选择比较了不同比例的乙腈-水对羟吡啶酮的提取效果,结果如图2所示,当提取溶剂为10%和50%乙腈水时,杂质峰及目标化合物的色谱峰严重变形。这是由于10%和50%乙腈在Hilic柱上的洗脱能力强于流动相,可能存在溶剂效应使峰形展宽。另外,根据相关文献[11],由于乙腈-水体系极性更大,减弱了目标化合物的分子内氢键,不利于发生分子内质子转移,反而导致分子间二聚体发生质子转移形成新的化合物,该原因也会导致峰形展宽。而采用纯乙腈作为提取溶剂,进行阴性样品加标实验时,其回收率均在90%以上且羟吡啶酮的峰形对称。因此本文采用纯乙腈为提取溶剂。
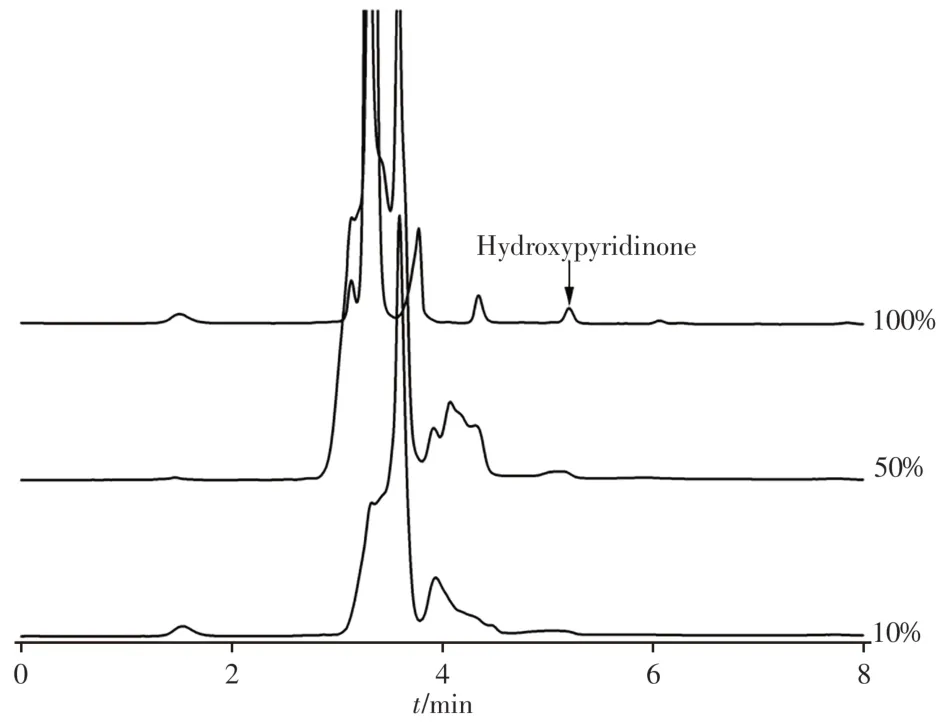
图2 不同含量乙腈提取的羟吡啶酮色谱图Fig.2 Chromatograms of hydroxypyridinone extracted with acetonitrile in different proportions
2.2.2 超声时间的优化超声提取具有操作方便、高效等特点,考察了超声时间对加标回收率的影响。将样品涡旋提取后,分别超声0、5、10、15、20 min,取样液进行分析。结果显示,随着超声时间的增加,目标化合物的峰面积有所增加,但超过15 min后,峰面积无明显增加,所以选择超声15 min进行提取。
2.3 线性方程及检出限
在优化条件下,对质量浓度为1.0 ~ 100.0 mg∕L的标准工作溶液进行测定,以目标物的峰面积(Y)对其质量浓度(X,mg∕L)进行线性回归分析,获得线性回归方程Y= 58.573X -85.464,结果表明羟吡啶酮在1.0 ~ 100.0 mg∕L质量浓度范围内线性关系良好,相关系数r= 0.999。对样品进行加标,以3倍信噪比计算方法检出限(LOD),以10倍信噪比计算方法定量下限(LOQ),羟吡啶酮的LOD和LOQ分别为8.0 µg∕g和20.0 µg∕g,可满足洗发水中羟吡啶酮的检测需求。
2.4 回收率及相对标准偏差
选取去屑洗发水阴性样品,分别添加40、100、400 mg∕kg 3个水平的羟吡啶酮标准工作溶液,按照本方法进行前处理和测定,每个水平平行测定6次,计算得到羟吡啶酮的平均回收率为91.8% ~96.7%,相对标准偏差(RSD)为1.3% ~ 3.7%。结果表明该方法有良好的回收率和精密度。
2.5 吡硫鎓锌中羟吡啶酮的测定及来源分析
对9个不同批次的吡硫鎓锌原料使用上述方法进行测定,结果有8批次检出羟吡啶酮,含量范围为28.3 ~ 87.2 mg∕kg。由于羟吡啶酮与HOPO为同一种化合物的互变异构体,且羟吡啶酮是较稳定的一种存在方式[1],为进一步探究吡硫鎓锌中测得目标物的形态,分别购买IL和Macklin两个品牌的羟吡啶酮标准品,Aladdin和Mackin两个品牌的HOPO标准品,进行对比实验。
用乙腈分别配制羟吡啶酮与HOPO的标准溶液,并分别对空白洗发水样品进行相同浓度加标实验,采用上述方法分别对标准溶液与提取样液进行分析。结果显示,两种化合物的保留时间及光谱图一致。
使用傅里叶变换红外光谱对4个标准品进行鉴别,如图3所示,1 626 cm-1处的吸收峰为羰基C= = O的伸缩振动,同时羰基大幅度红移;1 626 cm-1和1 508 cm-1之间的弱峰为C= = C共轭双键的伸缩振动,1 508 cm-1处为—N—O—的伸缩振动,3 075、3 040 cm-1处为C—H的伸缩振动。而3 650 ~ 3 200 cm-1处未发现OH的强吸收峰,说明化合物存在分子内氢键;718、752 cm-1处为C—H的弯曲振动吸收峰。羟吡啶酮与HOPO标准品红外谱图的主要特征吸收峰波长基本一致,但吸光强度有所差异,说明标准品中发生了互变异构,两种异构体共存,且互变异构体可能主要以酮式存在。该结果与文献[12]报道的互变异构过程相符。
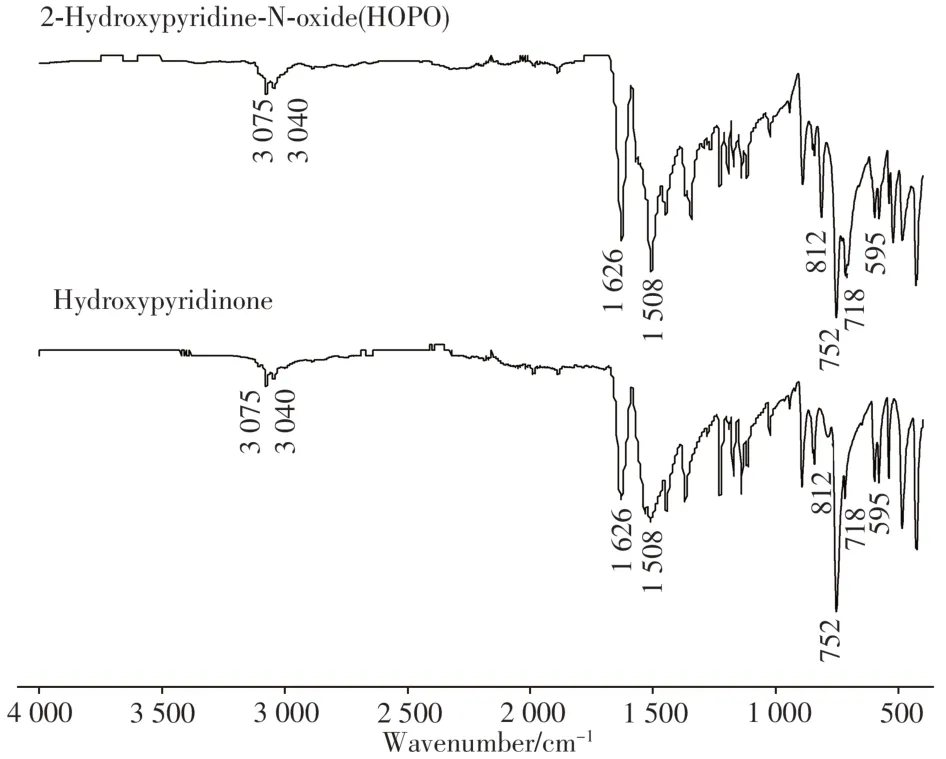
图3 羟吡啶酮和HOPO的红外光谱Fig.3 FTIR spectra of hydroxypyridinone and HOPO
进一步查阅资料发现吡硫鎓锌与HOPO的合成路线相似[13-15],如使用2-氯代吡啶为原料,用H2O2氧化生成N-氧化-2-氯吡啶,在碱性条件下巯基化后再与ZnSO4螯合即可生成吡硫鎓锌。该合成路线与文献[16]所报道的HOPO的制备方法十分相似。因此在吡硫鎓锌合成过程中极易生成HOPO杂质。
对比两种形态的标准品无法对两者进行有效区分,而红外光谱结果表明两者均以更稳定的羟吡啶酮形态存在,推测从原料中检出的羟吡啶酮很可能由HOPO互变产生。
2.6 实际样品测定
采用该方法对市面上采集的20个批次的去屑洗发水进行测定,结果在14个批次产品中检出羟吡啶酮,含量范围为16.8 ~ 120.2 mg∕kg。阳性样品的色谱图如图4所示。样品中的杂质峰未对目标物造成干扰,说明该方法能准确对去屑洗发水中的目标物进行定性定量分析。而检出羟吡啶酮的洗发水中均添加了吡硫鎓锌,因此羟吡啶酮极有可能是作为吡硫鎓锌原料中的杂质引入的。
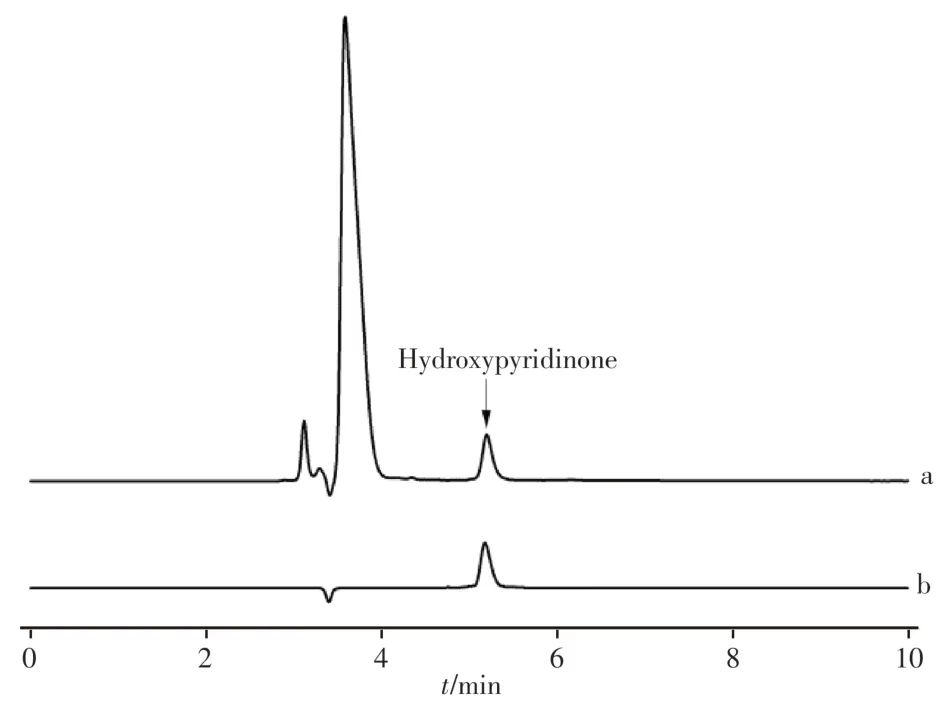
图4 羟吡啶酮的色谱图Fig.4 Chromatograms of hydroxypyridinone
3 结 论
通过优化样品前处理条件、色谱分离条件和质谱确证条件,建立了一种原料及洗发露中羟吡啶酮的测定方法,该方法前处理简单,快速、高效、准确,能够为去屑洗发露及吡硫鎓锌原料的质量监管提供有效的技术支持。该文首次报道了洗发露中禁用成分羟吡啶酮的添加情况及带入原因,为相关监管机构及生产企业提供了重要的风险线索。目前欧盟已将吡硫鎓锌列入禁用名单,我国《化妆品安全技术规范》(2015年版)规定吡硫鎓锌在去屑淋洗类发用产品中的使用限量为1.5%,在驻留类发用产品中的使用限量为0.1%[7],故羟吡啶酮仍然存在引入风险。考虑到羟吡啶酮的潜在基因毒性,呼吁相关部门重新评价吡硫鎓锌作为去屑剂使用的安全性,并对吡硫鎓锌原料加强监管,以保护广大消费者的健康。
