高效液相色谱-串联质谱法测定水产品中9种禁用染料类药物残留
2023-01-24孙良娟李红权蔡润斌庄姜云唐庆强唐媛媛
孙良娟,李红权,蔡润斌,庄姜云,唐庆强,唐媛媛,钟 键,黄 武*
(1.湛江海关技术中心,广东 湛江 524001;2.福州海关技术中心,福建 福州 350000)
亮绿(BG)又名碱性绿、灿烂绿,与孔雀石绿(MG)、结晶紫(CV)及其代谢产物隐性孔雀石绿(LMG)、隐性结晶紫(LCV)同属于碱性三苯甲烷类染料,工业上常用作着色剂,在水产养殖业中常用作鱼类水霉病、寄生虫病的治疗药物[1]。但此类化学物质具有明显的致畸、致癌、致突变的生物毒性。针对孔雀石绿和结晶紫及其氧化产物,多数国家和地区已严禁其用于水产养殖,并规定水产品中不得检出此类物质,我国农业农村部公告第250号也将其列入《食品动物中禁止使用的药品及其他化合物清单》,禁止其用于食用动物的养殖过程,但该类药物的非法使用仍时有发生[2-5]。而关于亮绿目前国内尚无相关检测标准与限量标准。
亚甲基蓝(MB)又名碱性湖蓝、美蓝,是一种人工合成的噻嗪类染料,在溶液中以离子型化合物存在,可通过与微生物酶系统竞争氢离子,使酶处于无活性状态,达到消毒杀菌目的,在水产养殖上常用来替代孔雀石绿、结晶紫,用于防治水霉病、寄生虫病等鱼类疾病,也作为抗真菌药物和水体消毒剂用于活鱼运输,提高鱼类的生存率[6-7]。亚甲基蓝有天青A(AZA)、天青B(AZB)和天青C(AZC)3种代谢物。与孔雀石绿和结晶紫相比,亚甲基蓝的毒性低,在孔雀石绿和结晶紫被禁用后,亚甲基蓝成为较好的替代品,但当其使用浓度超过安全范围时,仍会引起水生动物大量死亡,且高浓度的养殖废水会造成环境污染。研究表明,亚甲基蓝对鳑鲏鱼幼鱼的安全浓度为0.96 mg∕L[8],对三角鲂的安全浓度为1.24 mg∕L[9],对凡纳滨对虾的安全浓度为0.935 mg∕L[10]。目前,美国已禁止在食用水产动物中使用亚甲基蓝,日本《肯定列表》规定亚甲基蓝在水产品中的最高残留限量为10 µg∕kg[11],而国内尚未有相关明确规定。
水产品中三苯甲烷类兽药的检测方法主要有胶体金法[12-14]、酶联免疫法[15-17]、液相色谱法[18-19]以及液相色谱-质谱联用法[20-22]等,但同时检测三苯甲烷类与噻嗪类药物的文献报道较少。杨方等[23]采用液相色谱-质谱联用法对水产品中亚甲基蓝及3种代谢物进行检测,但未检测孔雀石绿、结晶紫、亮绿。余玮玥等[24]建立了同时检测水产品中亮绿、亚甲基蓝及代谢物的液相色谱-串联质谱法,但未包括孔雀石绿和结晶紫。侯建波等[25]采用液相色谱-质谱联用法同时检测水产品中的三苯甲烷类和噻嗪类药物,采用MCAX柱固相萃取净化,使用2种洗脱液分别洗脱三苯甲烷类和噻嗪类药物,洗脱过程繁琐且耗时长。本研究在参考相关文献的基础上,采用盐析萃取和固相萃取的净化方式,建立了检测水产品中三苯甲烷类和噻嗪类共9种药物的高效液相色谱-串联质谱法。该方法灵敏度高,适用性强,准确性好,可用于水产品中主要禁用染料类药物的残留检测和风险监控,以确保水产品质量安全。
1 实验部分
1.1 仪器与试剂
SCIEX Qtrap 4500液相色谱-串联质谱仪(SCIEX公司);Turbo Vap LV型氮吹浓缩仪(Zymark公司);T18型均质器(IKA公司);3-18K型高速冷冻离心机(西格玛公司);TDL-5-A型低速离心机(上海安亭科学仪器厂);Multi Reax涡旋振荡器(Heidolph公司);AL204-IC型分析天平(梅特勒公司);Waters XSelect HSS T3 液相色谱柱(2.1 mm × 100 mm,3.5 µm,Waters公司);Phenomenex Luna C18色谱柱(2.1 mm × 50 mm,5 µm);Waters Atlantis® T3柱(2.1 mm × 100 mm,3 µm);MCX萃取柱(6 mL∕500 mg,美国Waters公司);丙磺酸(PRS)固相萃取柱(3 mL∕500 mg,广州信谱徕公司);HR-XC阳离子柱(3 mL∕300 mg,北京振翔公司);滤膜(0.22 µm,上海安谱公司)。
亮绿、天青A(100 µg∕mL,天津阿尔塔公司);亚甲基蓝(97.1%)、天青B(88.8%)、天青C(51.9%)(天津阿尔塔公司);孔雀石绿及代谢物、结晶紫及代谢物混合标准溶液(100 µg∕mL,天津阿尔塔公司);乙腈、甲醇(色谱纯,Fisher公司);甲酸(色谱纯,Aladdin公司);乙酸铵(色谱纯,迪马公司);盐酸羟胺、对甲苯磺酸(分析纯,国药集团化学试剂有限公司),氯化钠(分析纯,广州化学试剂厂);洗脱液为乙腈-50 mmol∕L乙酸铵(pH 4.5,体积比1∶1)。
1.2 标准溶液的配制
分别称取适量的亚甲基蓝(97.1%)、天青B(88.8%)、天青C(51.9%)标准品,用甲醇溶解配制成质量浓度为100 µg∕mL的储备液,于-18 ℃保存;准确移取一定体积的100 µg∕mL上述9种药物储备液,用乙腈逐级稀释成质量浓度为50 µg∕L的混合标准工作液,于4 ℃保存,现用现配。
1.3 样品前处理
1.3.1 提 取待测样品取可食部分,搅碎混匀,制成肉糜状样品,装入洁净样品袋中,于-18 ℃冰箱冷冻保存。准确称取肉糜状样品3.0 g(精确至0.01 g),加入1 mL 0.25 g∕mL的盐酸羟胺、2 mL的0.5 mol∕L对甲苯磺酸、2 mL的0.1 mol∕L乙酸铵(pH 4.5),涡旋振荡5 min,使样品均匀分散,加入10mL乙腈、4 g氯化钠,剧烈振荡15 min,40 ℃超声提取10 min。在4 ℃下以12 000 r∕min离心10 min,移取上清液至氮吹管中,再加入10 mL乙腈重复提取1次,合并提取液。
1.3.2 净 化提取液在40 ℃下氮气吹扫浓缩至2 mL左右,加入4 mL经乙腈饱和的正己烷,涡旋振荡1 min,4 000 r∕min离心10 min,移取下层乙腈层至PRS固相萃取柱(预先用2 mL乙腈活化),再向氮吹管中加入2 mL乙腈重复上述步骤。待样液流尽后,用1 mL水和1 mL乙腈淋洗并减压抽干萃取柱,用6 mL洗脱液洗脱,收集洗脱液于带刻度玻璃试管中,于40 ℃氮吹至2 mL左右,用流动相定容至3 mL,过0.22 µm滤膜后上机测定。
1.4 仪器条件
色谱柱:Luna C18液相色谱柱(2.1 mm × 50 mm,5 µm),柱温:35 ℃,流速:0.35 mL∕min,进样量:10 µL。流动相:A为5 mmol∕L乙酸铵(含0.1%甲酸),B为乙腈。洗脱梯度为:0 ~ 2.0 min,5% ~95% B;2.0 ~ 4.0 min,95% B;4.0 ~ 4.1 min,95% ~ 5% B;4.1 ~ 6.0 min,5% B。
离子源:电喷雾离子源,扫描方式:正离子模式,监测模式:多反应监测(MRM)模式,电离电压为5 500 V,离子源温度为550 ℃,雾化气压力为345 kPa,辅助加热气压力为380 kPa,气帘气(N2)压力为207 kPa。9种目标化合物的质谱参数见表1。

表1 多反应监测模式下9种目标化合物的质谱参数Table 1 MS parameters of 9 target compounds in MRM mode
2 结果与讨论
2.1 质谱参数的优化
将甲醇稀释的1 µg∕mL的9种目标化合物混合标准溶液,用蠕动泵以0.01 mL∕min流速注入电喷雾离子源中,在持续的恒流状态下进样,优化各待测物的质谱参数。9种待测物均为离子型化合物,在正离子模式下以[M]+形式存在,通过一级质谱扫描(Q1 scan)方式,获得各待测物的质谱图并确定各自的母离子质量数。通过二级质谱扫描即碎片离子扫描(Product ion scan)获得各待测物的碎片离子图,确定碎片离子的准确质量数。选取响应高、干扰小的碎片离子与母离子组成离子通道,尽量避免选取脱水峰。在MRM模式下,分别精确优化各待测物子离子的去簇电压(DP)、碰撞能量(CE)等参数,确定最佳质谱电压参数。进一步优化离子源的喷雾电压、电离温度、雾化气压力、辅助加热气压力等参数,使各待测物的灵敏度最高,确定最佳质谱条件如“1.4”所示。
2.2 色谱条件的优化
C18柱是目前采用液相色谱-串联质谱检测有毒有害物质常用的色谱分离柱,本实验分别考察了Luna C18柱(2.1 mm × 50 mm,5 µm)、XSelect HSS T3柱(2.1 mm × 100 mm,3.5 µm)、Atlantis® T3柱(2.1 mm × 100 mm,3 µm)对9种目标化合物的分离效果。结果显示,以甲醇-水体系作为流动相时,3种色谱柱均出现拖尾、峰形变宽的现象;以乙腈-水体系作为流动相时,9种化合物的峰形得到较大改善,可能与乙腈在反相色谱柱上的洗脱能力大于甲醇有关。因此选择乙腈作为有机相。流动相中加入甲酸可提高待测化合物的离子化效率,进而提高其响应值;乙酸铵作为缓冲盐,可起到改善峰形、提高分离度的作用,因此在水相中加入0.1%甲酸与5 mmol∕L乙酸铵进一步考察3种色谱柱对9种待测物的分离效果。结果表明,XSelect HSS T3柱对天青C的保留能力弱,峰形前延、不对称,隐性结晶紫的峰宽变宽,响应值低;Atlantis® T3柱对亚甲基蓝与3种代谢物无法实现基线分离,且天青C的峰形不理想;Luna C18色谱柱可有效分离9种待测物,各组分的峰形尖锐、对称,响应好,灵敏度高,且天青C与隐性结晶紫的峰形明显改善。因此,选用Luna C18柱作为分离色谱柱。进一步优化梯度洗脱条件,使各组分的灵敏度最佳。图1为最优条件下10µg∕L的9种目标化合物混合标准溶液的MRM图。
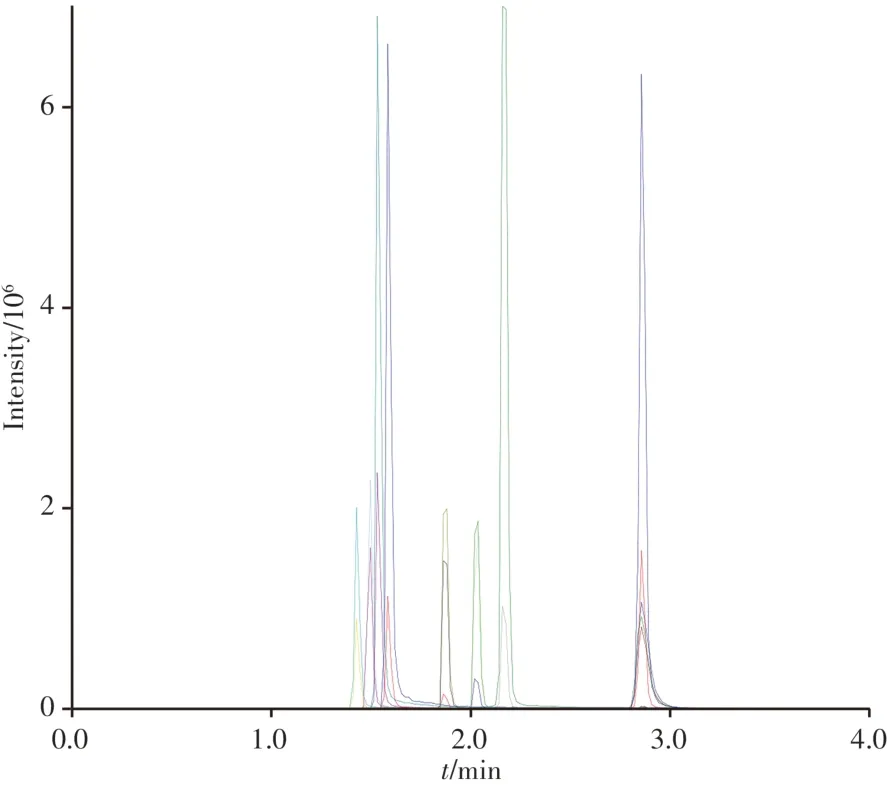
图1 最优条件下9种目标化合物的MRM图(10 µg∕L)Fig.1 MRM chromatograms of 9 target compounds under optimal conditions(10 µg∕L)
2.3 前处理条件的优化
2.3.1 液液萃取方式的选择文献[23-25]采用乙腈-0.1 mol∕L乙酸铵(pH 4.5)缓冲溶液作为提取剂,同时加入盐酸羟胺溶液防止孔雀石绿、结晶紫被氧化,加入对甲苯磺酸溶液与9种离子型待测物结合成离子对,以提高提取效率。但由于加入多种盐溶液,导致提取液中含有大量水,极大地影响了浓缩效率和效果;为除去水分需在提取液中加入二氯甲烷,导致萃取液体积大,浓缩时间长,操作繁琐且耗时。
在农兽药残留分析中,盐析萃取法是常用的除水方法。因此,考察了在提取液中加入氯化钠的除水效果。样品加标实验结果表明,当加入足量氯化钠并充分振摇离心后,水相与乙腈分层明显,上层乙腈层溶液澄清透明,9种待测物的回收率均在80%以上。同时,氯化钠的盐析作用还能促使提取的蛋白质等杂质在水相中沉淀,减少提取液中的有机杂质,提高待测物的响应值。本实验选择在提取液中加入约4 g氯化钠,使水相达到饱和,利用盐析萃取作用,实现了水相和有机相的分离且效果良好。
2.3.2 固相萃取柱的选择9种待测物均为自身带电荷的阳离子型化合物,使用具有阳离子交换吸附性能的萃取柱更有利于提高待测物的吸附净化效率。混合型阳离子交换柱(MCX)是以高度交联的聚苯乙烯和二乙烯基苯为基质、表面键合磺酸基的阳离子交换吸附柱;丙磺酸固相萃取柱(PRS)是以硅胶为基质、表面键合丙磺酸的强阳离子交换吸附柱;HR-XC阳离子柱的填料性质与MCX柱类似。上述3种萃取柱对阳离子型化合物均有良好的吸附保留能力,因此考察了其对9种化合物的吸附净化效果。移取1 mL 50 µg∕L的9种化合物混合标准溶液于3支玻璃管中,各加入2 mL按照“1.3.1”处理得到的罗非鱼空白基质提取液,混匀后转移至上述3种用乙腈活化的萃取柱中,用2 mL乙腈洗涤玻璃管2次并依次过柱。洗脱液为乙腈-50 mmol∕L乙酸铵(pH 4.5,1∶1)。结果表明,MCX柱和HR-XC柱对9种化合物的吸附作用较大,洗脱效果差,回收率低,两柱的回收率分别为69.8% ~ 82.7%和69.2% ~78.3%;PRS柱的回收率较为理想,9种化合物的回收率为82.3% ~ 93.7%。因此,选择PRS柱作为固相萃取净化柱。
2.3.3 洗脱液的选择文献[24]采用乙腈-0.1 mol∕L乙酸铵(pH 4.5,1∶1)作为PRS柱的洗脱液,并将洗脱液浓缩定容至3 mL后上机测定,但浓缩后上机液中乙酸铵的浓度增大,过高浓度的盐溶液增加了质谱仪污染的风险。文献[26]采用相同的洗脱液,将洗脱液稀释10倍后直接上机测定,由于三苯甲烷类与噻嗪类药物均为禁用药物,监控要求不得检出,此种方式不利于提高方法的灵敏度。本实验考察了pH值均为4.5的25、50、75、100 mmol∕L的乙酸铵溶液与乙腈等体积混合作为洗脱液的洗脱效果。移取1 mL 50 µg∕L的9种化合物混合标准溶液加入用乙腈活化的PRS萃取柱中,分别用上述4种洗脱液进行洗脱。由于9种待测物均为染料类药物,混合标准溶液呈蓝绿色,上柱后可被填料吸附形成色带。加入洗脱液后,待测物形成的色带在柱填料的移动速度随着乙酸铵浓度的增加而增快,表明洗脱液的洗脱能力随着乙酸铵浓度的增大而增强,所需洗脱时间随之缩短,洗脱液用量随之减少。按照乙酸铵浓度由高到低顺序,待测物色带完全脱离萃取柱所需的洗脱液体积依次为4、5、6、10 mL。综合考虑,实验选择乙腈-50 mmol∕L乙酸铵(pH 4.5,1∶1)作为洗脱液,9种待测物的回收率在78.4% ~ 97.1%范围内,可满足方法学要求。
2.4 方法学验证
2.4.1 基质效应水产品样品成分复杂,虽然提取液经过净化处理,但仍有部分杂质可能与待测物随流动相进入质谱离子源,对目标化合物的离子化效果产生影响,进而导致基质效应(ME)。采用下式计算基质效应:ME = (1-基质匹配标准曲线的斜率∕溶剂标准曲线的斜率)× 100%[27]。分别以空白罗非鱼样品提取液和乙腈-0.1%甲酸溶液(2∶8,体积比)配制标准曲线溶液并上机测定,得到9种化合物的ME为31.8% ~ 76.4%,表明存在基质抑制效应。因此采用空白基质匹配法制作标准曲线,以抵消基质效应,提高定量结果的准确性。
2.4.2 线性范围、检出限与定量下限准确移取适量混合标准工作溶液,用空白基质提取液稀释成标准工作曲线溶液,以各待测化合物的质量浓度(x,µg∕L)为横坐标,各化合物定量离子的色谱峰面积(y)为纵坐标拟合标准曲线。以空白罗非鱼作为样品基质进行加标实验,加标浓度为0.5、1.0、2.0、5.0、10.0 µg∕kg,采用本方法进行前处理和检测,以定量离子色谱峰的信噪比S∕N≥ 3且满足方法准确度要求时的最低浓度为方法检出限(LOD),以S∕N≥ 10对应的最低浓度为定量下限(LOQ)。结果显示,9种待测物均在0.5 ~ 20.0 µg∕L范围内呈良好线性关系(r2> 0.99),LOD均为0.5 µg∕kg,LOQ均为1.0 µg∕kg(见表2)。

表2 9种目标化合物的线性范围、线性方程、相关系数、检出限与定量下限Table 2 Linear ranges,linear equations,correlation coefficients(r2),limits of detection(LODs) and limits of quantitation(LOQs) of 9 target compounds
2.4.3 回收率与相对标准偏差为进一步验证方法的可行性与适用性,分别对空白罗非鱼、金鲳鱼、南美白对虾和青蟹样品在0.5、1.0、5.0 µg∕kg水平下进行了加标回收率和精密度实验(n= 7)。如表3所示,9种待测物在上述样品基质中的平均回收率为76.8% ~ 104%,相对标准偏差(RSD)为4.3% ~10%,表明方法的准确性和重复性良好,能满足分析检测要求。

表3 9种目标化合物在4种样品中的平均回收率和相对标准偏差(n = 7)Table 3 Average recoveries and RSDs of 9 target compounds spiked in four samples(n = 7)
2.5 实际样品检测
采用本方法对实验室留存的海鲈鱼、石斑鱼、罗非鱼和斑节虾共16个样品进行检测,均未检出目标化合物。对健康罗非鱼(体质量200 ~ 300 g)进行亚甲基蓝药浴实验,药浴质量浓度约为1 mg∕L,4 h后采样,取肌肉部分按照本方法进行测定。罗非鱼肌肉中检出亚甲基蓝和天青B,含量分别为3.41、1.89 µg∕kg,未检测到天青A和天青C。图2为检出亚甲基蓝与天青B的样品的MRM图。数据表明,本方法准确可靠,可操作性强,能够满足水产品中三苯甲烷类与噻嗪类药物残留的检测要求。
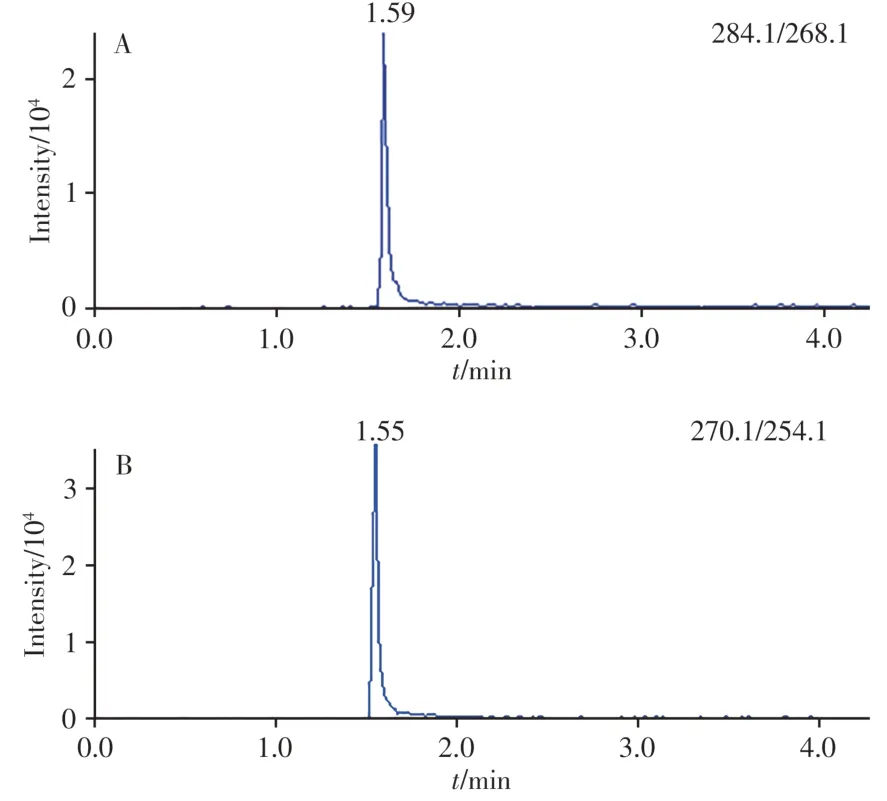
图2 罗非鱼阳性样品中亚甲基蓝(A)和天青B(B)的MRM图Fig.2 MRM chromatograms of methylene blue(A) and azure B(B) in tilapia positive samples
3 结 论
本文建立了检测水产品中三苯甲烷类与噻嗪类共9种禁用染料类药物的高效液相色谱-串联质谱法,样品经乙腈-0.1 mol∕L乙酸铵(pH 4.5)提取后,采用盐析萃取、正己烷液液萃取和固相萃取的净化方式,净化效果良好,有效去除了干扰,提高了灵敏度。在优化的实验条件下,9种待测物在0.5 ~ 20.0 µg∕L范围内线性关系良好,检出限为0.5 µg∕kg,定量下限为1.0 µg∕kg,样品加标回收率为76.8% ~ 104%,RSD为4.3% ~ 10%。本方法定量准确,灵敏度高,结果稳定可靠,可用于水产品中禁用染料类药物的日常检测和残留监控。
