黑守瓜中一种新negevirus的鉴定和分析
2023-01-19王海强张岩叶庄新卓继冲张传溪李俊敏陈剑平
王海强,张岩,叶庄新,卓继冲,张传溪,李俊敏,陈剑平
(宁波大学植物病毒学研究所,农产品质量安全危害因子与风险防控国家重点实验室/农业农村部和浙江省植物保护生物技术重点实验室,浙江 宁波 315211)
negevirus(nege-like virus)是 一 类 正 义 单 链RNA病毒,在自然界中广泛分布,主要侵染昆虫纲、甲壳纲等节肢动物,是近几年新增的一个病毒分类单元。negeviruses 的基因组一般由9 000~10 000个核苷酸(nucleotide, nt)组成,包含至少3 个开放阅读框(open reading frame, ORF)。ORF1 最长,靠近病毒基因组的5′末端,一般包含4 个保守结构域:病毒甲基转移酶(viral methyltransferase, vMet)结构域、RNA 核糖体甲基转移酶(RNA ribosomal methyltransferase, FtsJ)结构域、RNA 病毒解旋酶(viral helicase, Hel)结构域以及RNA 依赖的RNA聚合酶(RNA-dependent RNA polymerase, RdRp)结构域。ORF2 和ORF3 则分别编码糖蛋白DISBORF2_chro 和膜蛋白SP24[1-2]。系统发育分析结果表明,negeviruses 在进化过程中形成了2 个不同的分支,分别为nelorpivirus和sandewavirus[3]。前人的研究表明,部分昆虫中negeviruses 的ORF1 不编码FtsJ,还 有 一 些 新 鉴 定 的negeviruses 的ORF1 和ORF2存在重叠区域[4-7]。
在昆虫细胞中,RNA病毒的自我复制会诱发宿主的RNA 干扰抗病毒免疫反应,并产生靶向RNA病毒基因组的病毒来源的干扰小RNA(virusderived small interfering RNA,vsiRNA)。vsiRNA由昆虫宿主的Dicer 酶切割产生,其长度主要为21~22 nt,而且vsiRNA 的5′末端核苷酸常为腺嘌呤(adenine, A)和尿嘧啶(uracil, U)[8]。不同长度的vsiRNA在病毒基因组的分布不具有链特异性,但是会形成明显的热点区域[8]。negeviruses 侵染双翅目的粪蝇、半翅目的蚜虫及粉虱等多种昆虫后,均能在宿主体内产生大量vsiRNA,且长度偏好性、正负义链比例及5′末端核苷酸分布等均具有典型昆虫宿主Dicer酶的切割特征,表明negeviruses在宿主细胞中的复制十分活跃[4,7,9]。
传统的RNA 病毒鉴定主要依赖于反转录聚合酶链反应(reverse transcription-polymerase chain reaction,RT-PCR)和血清学检测等方法,但这些方法主要针对已知病毒的鉴定[10]。对于未知的新病毒,之前主要是对病毒进行分离培养并通过电子显微镜观察完成鉴定,但这些方法具有局限性,且很难在较短时间内完成[10]。随着测序技术的发展,以第二代高通量测序技术为基础的病毒宏转录组技术为新病毒的鉴定提供了新方案,该技术可以一次性测定样品中所有的转录本,包括RNA病毒通过复制产生的全长基因组。基于宏转录组技术的新病毒鉴定方案,通过对测序数据的处理、分析,从而鉴定样品中的病毒序列,进一步结合Sanger 测序、cDNA 末端快速扩增技术(rapid amplification of cDNA ends,RACE)及系统发育分析等方法,完成新病毒的鉴定及全长基因组的获取。
黑守瓜(Aulacophora lewisii)是一种鞘翅目叶甲科的农业害虫,主要危害瓜类蔬菜。黑守瓜成虫在土壤中产卵,幼虫主要危害瓜类蔬菜根部,成虫则以瓜类蔬菜的叶、茎、花、果实为食[11]。2021年,本实验室利用宏转录组技术在黑守瓜中鉴定到一种新的昆虫特异性病毒Aulacophora lewisii iflavirus 1[12]。基于此,本研究继续以黑守瓜为试验对象,通过宏转录组技术鉴定并获得了黑守瓜中一种新的negevirus,测定该病毒的基因组全长,并对其基因组结构、分类地位及病毒来源的干扰小RNA(vsiRNA)等作出进一步分析。
1 材料与方法
1.1 昆虫样品采集和RNA 提取
黑守瓜采集于浙江省温州市某黄瓜田块(叶片),将单头黑守瓜活虫直接置于液氮中,并使用Trizol试剂盒(RNAiso Plus,日本TaKaRa公司)提取黑守瓜的总RNA(提取方法参考上述试剂盒说明书)。总RNA经质控检测合格后,分成3份,分别用于转录组测序、小RNA提取和测序、病毒序列验证。
1.2 转录组测序
黑守瓜总RNA 利用Illumina HiSeq 4000 平台进行转录组测序。原始序列数据(raw data)利用Trimmomatic 0.39 软件进行质控(去除低质量序列和接头序列),再利用Trinity 2.8.5 软件对质控后的数据进行无参考基因组组装(de-novo组装)[13],得到样品的转录组拼接序列(Contigs)。使用Trinity 2.8.5 提供的脚本(Trinity_stats.pl)统计转录本N50(Contig N50)以对转录组组装质量进行评估。
1.3 昆虫宿主物种鉴定
利用BLASTn工具将拼接好的转录组数据与生命条形码数据库(Barcode of Life Data System,BOLD;https://www.boldsystems.org/)进行同源性比对。将比对上的昆虫线粒体细胞色素氧化酶亚基Ⅰ(cytochrome oxidase subunitⅠ,COⅠ)基因与NCBI(https://www.ncbi.nlm.nih.gov/)中非冗余核酸库(Non-Redundant Nucleotide Sequence Database,NT)进行比对,以明确和验证具体的昆虫物种。
1.4 黑守瓜中新病毒的鉴定
下载NCBI 上所有病毒的最新蛋白序列信息(https://www.ncbi.nlm.nih.gov/genome/viruses)并构建本地病毒数据库,利用拼接好的转录本作为种子序列与该数据库进行同源性比对(DIAMOND BlastX 0.9.28.129软件)。将鉴定到的潜在病毒的Contigs序列同时与NCBI 的非冗余蛋白库(Non-Redundant Protein Sequence Database,NR)及NT库进行同源性比对以明确病毒序列。
1.5 病毒基因组确认及全长序列获取
根据转录组中鉴定到的新病毒Contigs序列设计特异性引物,引物序列见表1。使用预混液形式的2步法反转录聚合酶链反应试剂(去基因组)[HiScript®ⅡQ RT SuperMix for qPCR (+gDNA wiper)](购自南京诺唯赞生物科技股份有限公司),利用黑守瓜的总RNA反转录合成cDNA,并以此为模板进行聚合酶链反应(PCR)。使用1%琼脂糖凝胶对PCR产物进行电泳检测,利用凝胶DNA 小量回收试剂盒[天根生化科技(北京)有限公司]进行回收、连接转化,产物送至杭州有康生物科技有限公司进行Sanger 测序。使用RACE 试剂盒(SMARTer®RACE 5′/3′ Kit,日本TaKaRa 公司)通过cDNA 末端快速扩增技术(RACE)克隆新病毒基因组5′末端和3′末端的缺失序列(RACE 引物序列见表1)。根据RACE 以及Sanger 测序得到不同序列的重叠区,利用多序列比对方法对其进行拼接,以得到完整的新病毒基因组序列。
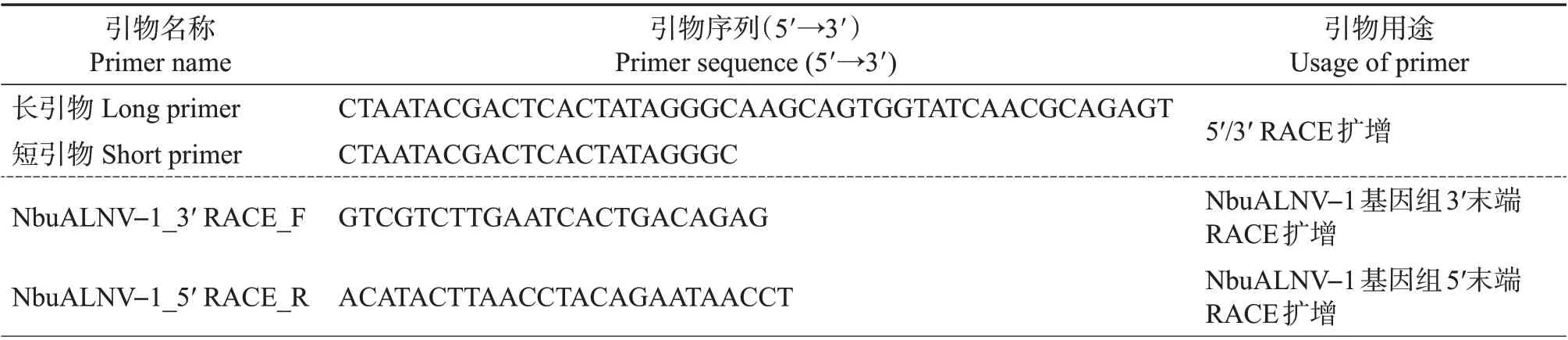
表1 引物信息Table 1 Primer information
1.6 病毒基因组序列分析
利用Expasy 在线网站(https://web.expasy.org/translate)预测病毒基因组各个ORF,并在InterProScan网站(https://www.ebi.ac.uk/interpro/search/sequence)上注释不同ORF 所编码的相应蛋白。同时,使用Bowtie2 2.3.5.1 软件将高通量测序质控得到的原始读长(reads)回贴到明确的病毒基因组全长序列中,并利用R语言4.1.3对结果进行可视化,得到病毒基因组在宿主体内的表达丰度。

表1(续) Continuation of Table 1
1.7 系统发育分析
使用MAFFT 7.487 软件对新鉴定病毒的RdRp序列与已报道的negeviruses 的RdRp 序列进行多序列比对[14],利用ModelTest-NG软件对构建系统进化树的模型进行预测和评估[15],最后利用RAxML-NG软件基于最大似然法(maximum likelihood, ML)构建系统进化树,将进化树的自展值(bootstrap)设置为1 000[16]。
1.8 小RNA 分析
利用小RNA 文库构建试剂盒(Illumina TruSeq Small RNA Sample Preparation Kit)构 建 用 于 小RNA 测序的文库,将文库置于Illumina HiSeq 2500平台进行单端测序。数据下机后,对小RNA原始数据进行质控(去除低质量序列、接头序列等),再使用Bowtie 1.2.3软件将小RNA数据回贴到已明确的病毒全基因组中(不允许错配)。在此基础上参照先前报道的方法,使用Perl脚本对小RNA数据进行特征分析并对结果进行可视化[17]。
2 结果与分析
2.1 高通量测序与数据质控
田间采集到的疑似黑守瓜单头样品经转录组测序,共得到21 514 793 个原始读长(reads)。对测序数据进行质控,获得21 084 498 个高质量读长(clean reads),进一步使用Trinity 2.8.5软件进行denovo组装得到104 125 条拼接的转录本Contigs 序列。转录组组装预测基因的平均序列长度为1 700 nt,Contig N50为1 499 nt。
2.2 黑守瓜中一种新negevirus 的鉴定
将组装好的Contigs 在BOLD 数据库中进行同源性比对,提取对应的昆虫COⅠ序列并与在线的NCBI NT 库比对,发现该序列与已报道的黑守瓜COⅠ序列(NC_039712.1)有99.82%的序列同源性,明确了该昆虫物种为黑守瓜(A.lewisii)。
进一步利用拼接好的黑守瓜Contigs 与构建的本地病毒数据库作同源性比对,发现黑守瓜中有一个Contig(长度为8 585 nt)与已报道的nege-like virus 病毒Hangzhou merodon fulcratus virga-like virus 1(UHK03236.1)同源性最高(36.33%),表明该Contig可能是黑守瓜中一种新的negevirus。随后利用RT-PCR、RACE 及Sanger 测序明确了该病毒的基因组全长为9 832 nt,GC 含量为34.26%(附图1,http://www.zjujournals.com/agr/CN/10.3785/j.issn.1008-9209.2022.06.293)。我们将新鉴定的病毒命名为Nbu Aulacophora lewisii nege-like virus 1(NbuALNV-1)。NbuALNV-1 的基因组序列已上传至GenBank 数据库(登录号:ON442311)。
2.3 NbuALNV-1 序列分析
NbuALNV-1的基因组包含6个ORFs,其5′末端和3′末端各有1 个非翻译区(untranslated region,UTR),5′UTR 的长度为292 nt,3′UTR 的长度为77 nt(图1)。对NbuALNV-1不同ORF的保守结构域进行预测,结果表明:靠近病毒基因组5′末端的ORF1包含3 个保守结构域,即病毒甲基转移酶(vMet)结构域、RNA 病毒解旋酶(Hel)结构域和RNA 依赖的RNA 聚合酶(RdRp)结构域,ORF2 具有糖蛋白DISB-ORF2_chro 结构域,ORF4 则编码一个预测的膜蛋白SP24(图1)。分析结果表明,NbuALNV-1具有典型的negeviruses保守结构域,与先前昆虫中报道的结果[5]一致。为明确NbuALNV-1在黑守瓜中的丰度,将质控后的高质量读长(clean reads)回贴到NbuALNV-1基因组,结果表明,NbuALNV-1来源的读长(reads)大多聚集在病毒基因组的3′末端,而5′末端及其他位置的病毒丰度相对较低(图1)。
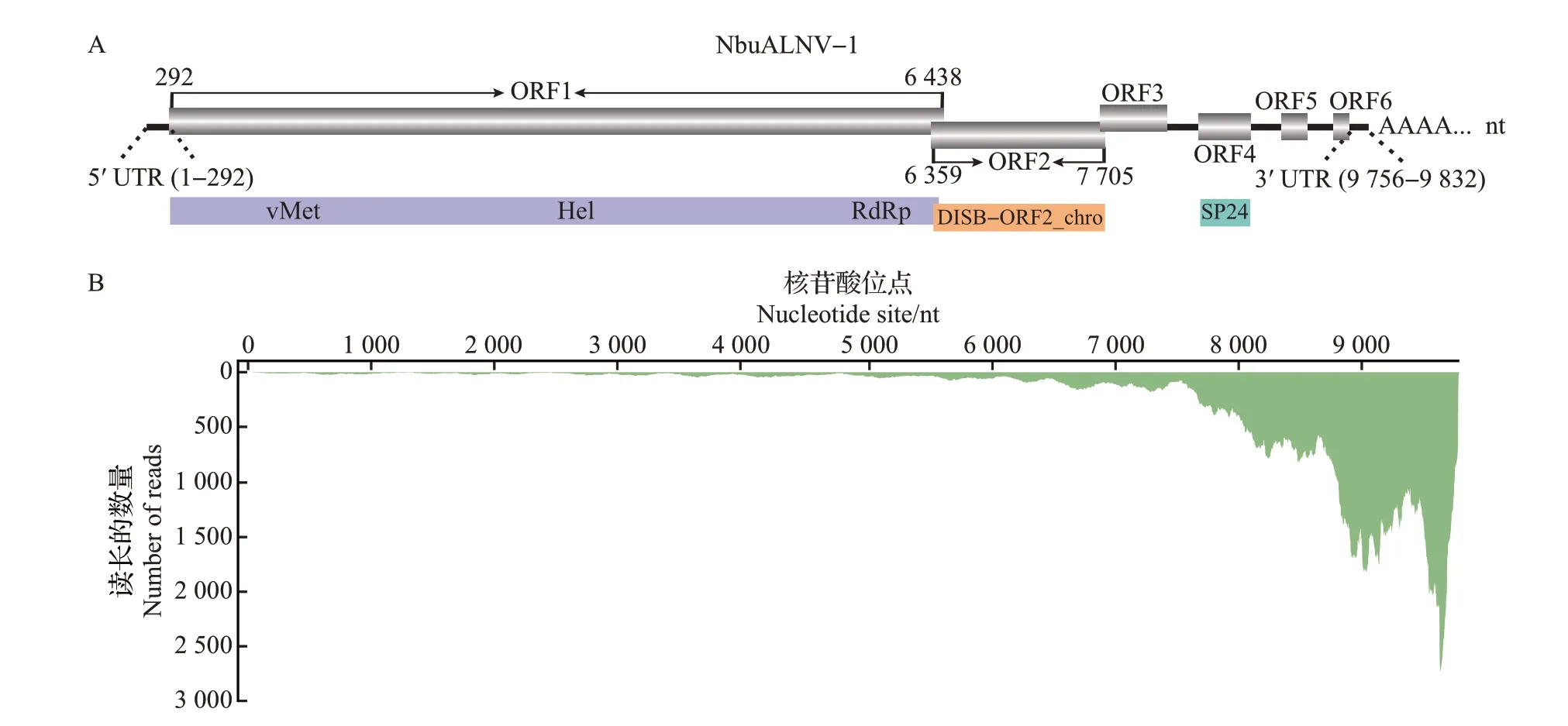
图1 NbuALNV-1的基因组结构与读长覆盖度Fig.1 Genome structure and reads coverage of NbuALNV-1
2.4 NbuALNV-1 与其他negeviruses 的同源性比对和分析
为了分析NbuALNV-1 与其他negeviruses 的同源性,使用NbuALNV-1的RdRp氨基酸序列与已报道的nege/nege-like virus 相应RdRp 序列进行同源性比对。结果(表2)表明,NbuALNV-1与部分已报道的nege/nege-like virus 的序列同源性为26.2%~35.8%。从图2 中可知,NbuALNV-1、nege/nege-like virus 及植物病毒北岛病毒科(Kitaviridae)的RdRp氨基酸序列中均含有保守的同源序列,包括3 个基序,模式分别为GDD(基序A)、DX(4-5)D(基序B)和GX(2-3)TX(3)N(基序C)。

图2 nege/nege-like virus与NbuALNV-1的RdRp氨基酸序列同源性比对分析Fig.2 Homology alignment analysis based on RdRp amino acid sequences of nege/nege-like virus and NbuALNV-1
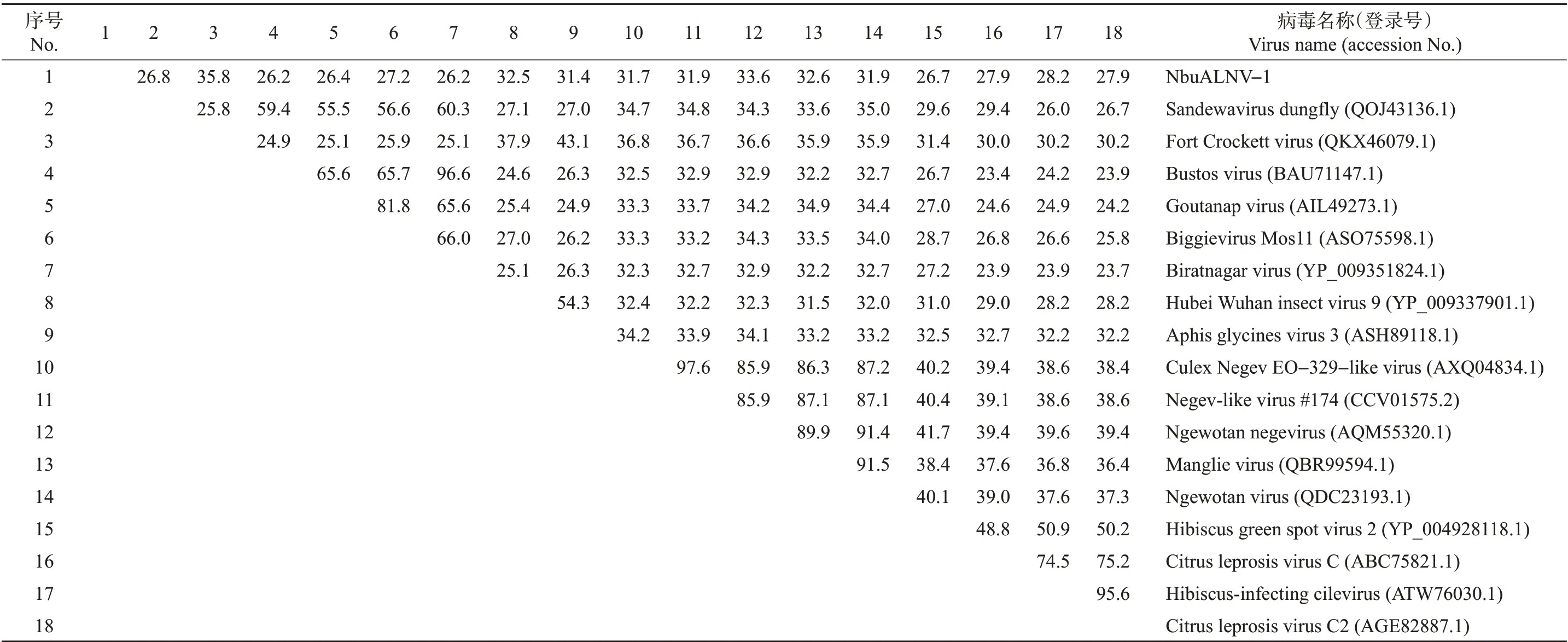
表2 NbuALNV-1与已报道的negeviruses RdRp氨基酸序列的同源性Table 2 Homology of RdRp amino acid sequences among NbuALNV-1 and reported negeviruses
2.5 NbuALNV-1 的分类地位
为进一步分析NbuALNV-1与之前报道的nege/nege-like virus 之间的系统发育关系,利用它们的RdRp 氨基酸序列进行同源性比对并构建系统进化树。结果表明:NbuALNV-1 并未与已报道的2 个negeviruses 分支类群(sandewavirus 和nelorpivirus)聚合在一起,而是与另外3 种昆虫特异性病毒(Hubei Wuhan insect virus 9、Aphis glycines virus 3和Fort Crockett virus)聚合,形成了一个独立分支(unclassified)。值得一提的是,该分支与植物病毒北岛病毒科(Kitaviridae)的病毒聚合在一起(图3),表明昆虫病毒和植物病毒在进化上具有较近的亲缘关系。
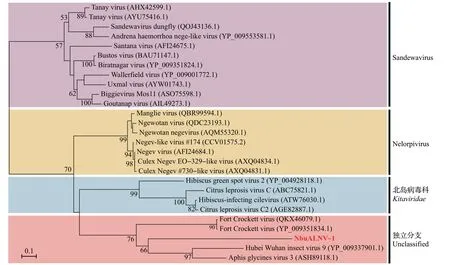
图3 已报道的nege/nege-like virus与NbuALNV-1的RdRp氨基酸序列构建的系统发育进化树(基于最大似然法)Fig.3 Phylogenetic tree of RdRp amino acid sequences of NbuALNV-1 and reported nege/nege-like virus based on the maximum likelihood
2.6 NbuALNV-1 来 源 的 干 扰 小RNA 分 析
本研究进一步对NbuALNV-1病毒来源的干扰小RNA(vsiRNA)进行分析,通过小RNA 测序共获得了2 640 965 个小RNA 序列,其中6 622 个为NbuALNV-1 来源的vsiRNA。进一步的分析结果表明,NbuALNV-1 产生的vsiRNA 长度主要为21 nt,且源于病毒基因组正义链及其互补链(负义链)的vsiRNA 数量基本一致(图4A)。这些vsiRNA 在NbuALNV-1 基因组的各个位置均匀分布,且无明显的正负义链偏好性(图4B)。此外,NbuALNV-1来源的vsiRNA 5′末端呈现出典型的腺嘌呤(A)和尿嘧啶(U)富集现象(图4C)。这些结果均表明NbuALNV-1侵染黑守瓜后能成功诱发宿主基于小RNA的RNA干扰抗病毒免疫反应。
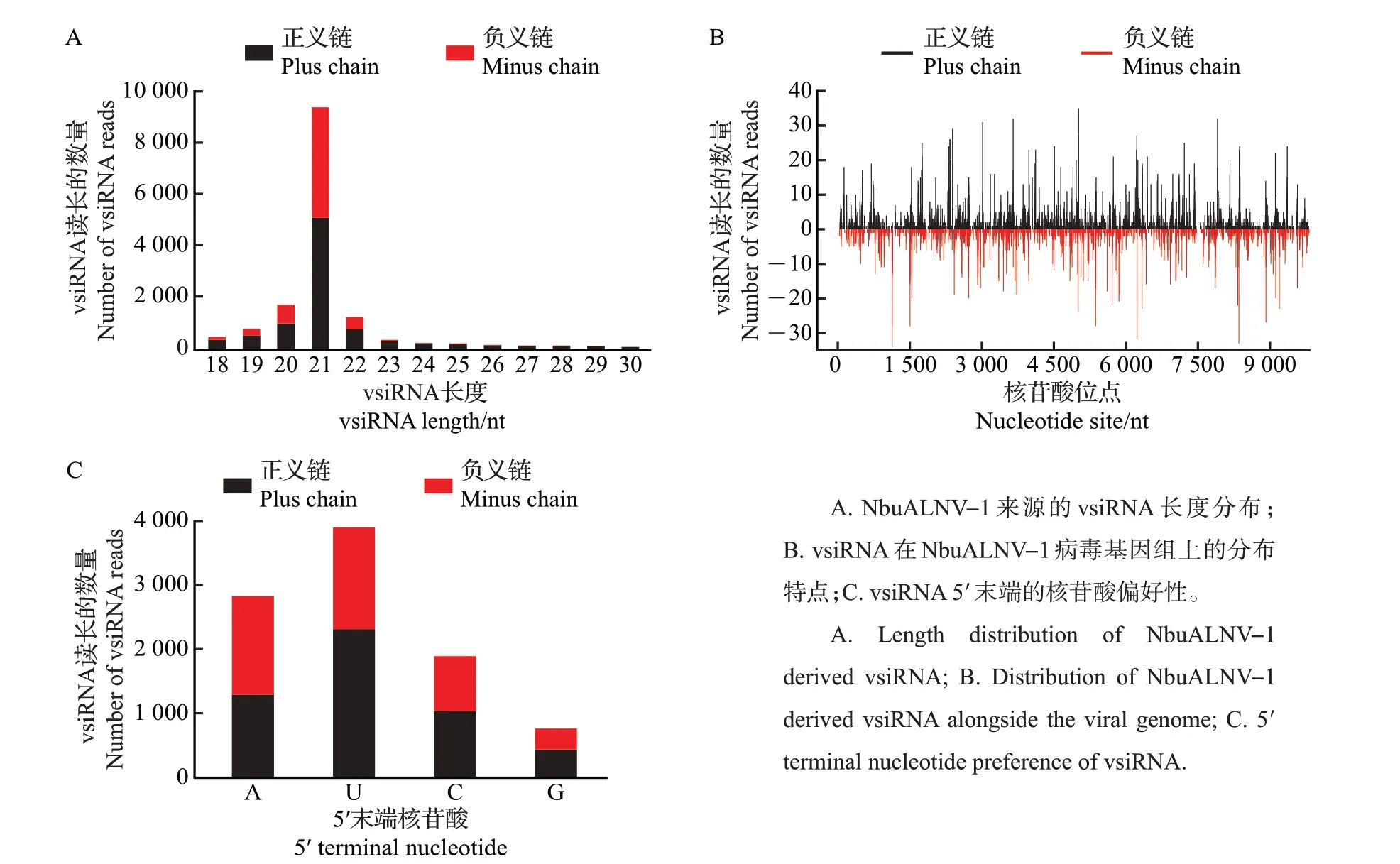
图4 NbuALNV-1来源的vsiRNA分析Fig.4 Analysis of NbuALNV-1 derived vsiRNA
3 讨论
基于高通量测序技术的病毒宏转录组为新病毒的鉴定提供了一种快速、高效的方法。本研究利用病毒宏转录组技术从黑守瓜中鉴定到一种新的negevirus(NbuALNV-1),其全基因组大小为9 832 nt,具有6个ORFs且包含多个negeviruses保守结构域。目前在其他昆虫中鉴定到的negeviruses 一般包含3~4 个ORFs,表明自然界中negeviruses 的基因组结构存在多样性[1,6]。另外,部分已报道的negeviruses ORF1中还具有一个FtsJ结构域,而黑守瓜中的NbuALNV-1 与蚜虫中的Indomegoura negelike virus 1类似,未鉴定到该保守结构域,因此其在negeviruses侵染和复制过程中的作用仍有待进一步研究和明确[2,4,6]。
系统发育分析表明,NbuALNV-1 与已明确的negeviruses的2个类群sandewavirus和nelorpivirus均未聚合在一起,而是与其他昆虫特异性病毒聚合形成一个新的独立分支,表明NbuALNV-1 可能是negeviruses中的一个新类群,同时,随着昆虫中新的negeviruses 类群不断被鉴定出来,可能还有更多类群需要进一步被明确。
基于干扰小RNA(small interfering RNA,siRNA)的抗病毒通路是节肢动物应对外源病毒侵染的主要免疫途径,该通路的激活在宿主体内伴随着大量vsiRNA的产生[17]。有关NbuALNV-1来源的siRNA分析结果显示,vsiRNA 呈现出典型的昆虫宿主Dicer 酶切割特点(21~22 nt 的长度偏好性、相近的正负义链比例及5′末端核苷酸的A/U 偏好性),表明NbuALNV-1成功激活了黑守瓜的siRNA抗病毒免疫系统。有意思的是,NbuALNV-1 来源的vsiRNAs 的长度偏好为21 nt,这种长度分布模式和半翅目(如蚜虫)中negeviruses 的vsiRNAs 不同(以22 nt 为主)[4],但和已报道的双翅目(粪蝇)中的vsiRNAs 一致[7]。这些结果表明,不同目昆虫中vsiRNAs 的切割机制可能存在差异,而鞘翅目中的siRNA抗病毒通路在外源病毒侵染过程中的具体机制还有待后续进一步研究。
综上所述,本研究通过宏转录组技术从黑守瓜中鉴定出一种新的negevirus,命名为NbuALNV-1,明确了其基因组全长为9 832 nt;并对NbuALNV-1的基因组结构特点、分类地位及产生的vsiRNAs 等特点进行了分析和研究。NbuALNV-1是在鞘翅目昆虫中发现的首个negevirus,该病毒的鉴定和分析有助于我们进一步了解昆虫中negeviruses 的多样性,为后续黑守瓜昆虫共生病毒的功能研究及其在防控方面的潜在应用提供了理论依据。
