4例CHARGE综合征新生儿的临床表现及CHD7基因突变分析
2023-01-17鲁艺李红易丽君段芳芳蒋文星胡清华
鲁艺,李红,易丽君,段芳芳,蒋文星,胡清华
(江西省儿童医院中心实验室,南昌 330006)
CHARGE综合征(OMIM 214800)是一种常染色体显性遗传疾病,发病率大约在1/8500~1/15000[1]。以眼部缺损、心脏疾病、后鼻孔闭锁、生长发育迟缓、泌尿生殖系统缺陷/性腺功能减退以及耳部畸形/耳聋为主要临床症状[2-3],肾脏异常、唇/腭裂和气管食管瘘等也被频繁报道[4]。CHD7基因是主要的致病基因,CHD7突变大约占总病例的60%[5],并且在严格符合Blake或Verloes诊断标准[5-6]的患者中有90%~95%携带CHD7变异体[6]。因此,2016年Hale等人[7]建议将CHD7基因变异加入到主要诊断标准中,致病性CHD7突变加一个主要表现即可诊断CHARGE综合征。随着分子检测技术的进步,越来越多的可疑病例可以及时得到基因检测[8-9],通过了解CHARGE综合征的不同基因型,可以为愈来愈多的CHD7突变体明确基因型与表型的关联;通过CHD7突变患儿临床表型的分析,亦可以扩大CHARGE综合征的临床表型。目前,国内CHARGE综合征报道相对较少,本文报道本院2018年至2019年收治的4例携带CHD7基因突变的患儿,对他们临床表现进行总结及CHD7基因突变进行分析,以增加临床医生对CHARGE综合征发展和临床表现的了解,并及时诊断,从而更及时地进行临床护理和获得基因咨询。
1 对象与方法
1.1 对象 回顾性病例总结。纳入2017年1月至2019年12月本院收治的4例(男1例、女3例)存在CHD7基因突变患儿为研究对象。正常组为265名新生儿常见遗传病基因筛查样本,患儿父母均签署知情同意书,研究获得医院伦理委员会批准(批准号:JXSETYY-YXKY-20180015)。
1.2 方法 高通量测序及父母Sanger验证:采集患儿及父母静脉血样,提取患儿基因组DNA,使用Tagment DNA enzyme酶 切法(Illumina)将DNA打断为100~200bp片段。采用IDTxGenExome V2全外显子试剂盒(IDT)对靶向20000+目标基因的外显子区域进行富集捕获。捕获文库经过Agilent 2100 Bioanalyzer Instrument(Agilent)质检,确认合格后采用Illumina Novaseq6000(Illumina)高通量测序仪测序。下机后测序原始数据以FastQC软件进行质控分析,后续进一步采用Sentieon(Sentieon)比对软件将测序序列与人类基因组参考序列(GRch37/hg19)比对并分析单核苷酸变异和插入/缺失,最后将VCF格式的变异文件导入至FLIMS系统(Fulgent Genetics),对变异位点根据致病性、表型和家系遗传模式进行筛选。通过ACMG标准对全部变异进行分级。而后提取先证者父母的外周血DNA,对二代测序中检测出的致病、可能致病与临床意义不明3类突变位点进行父母Sanger测序验证。
2 结果
2.1 二代测序结果及父母Sanger验证结果4例先证者CHD7基因分别存在c.686ldelC(p.Y2288Tf-sTer9)、c.7192C>G(p.R2398G)、c.1366C>T(p.Q456X)和c.5210+3A>G杂合变异(见图1)。Sanger测序验证结果显示4例患儿的父母均未携带上述变异,4例先证者变异均为新发突变(见图2)。c.686ldelC(p.Y2288TfsTer9)变异会导致所编码的氨基酸提前截短,影响蛋白的正常功能,查阅HGMD数据库、ExAC数据库及相关文献,该变异为未报道的新变异;c.7192C>G[10]、c.1366C>T[4]以及c.5210+3A>G[11]均已被报道过。在265名正常对照中未检测到以上变异。本文报道的4个突变位点的区域均位于高度保守区(见图3),故本文报道的变异区域在脊椎动物中可能具有重要作用,该区域的变更可能导致疾病的发生。
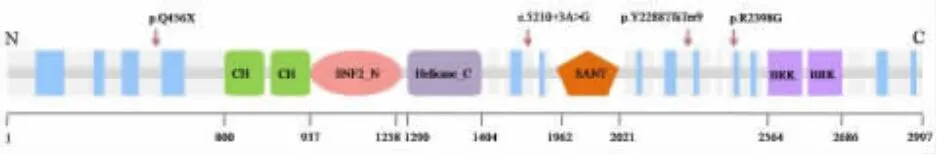
图1 CHD7蛋白结构示意图[12]CH:chromo结构域;SNF2:ATP酶或解旋酶结构域;Helicase_C:解旋酶结构域;SANT:SANT结构域;BRK:BRK结构域。红色箭头为本文4例患儿氨基酸突变位置。

图2 Sanger验证测序结果A,C和E分别为患儿1,患儿1父亲和患儿1母亲的测序图;B,D和F分别为患儿2,患儿2父亲和患儿2母亲的测序图;G,I和K分别为患儿3,患儿3父亲和患儿3母亲的测序图;H,J和L分别为患儿4,患儿4父亲和患儿4母亲的测序图。箭头示基因变异位点,4例先证者的父母均未检测到CHD7基因突变。

图3 不同种属间CHD 7蛋白序列保守性分析 蛋白质序列下面的(*)表示保守序列、(:)表示保守变体、(.)表示半保守变体、()表示非保守序列。红色箭头处分别为本文4例患儿突变位点。
2.2 临床诊断分析及临床症状特点 依据相关文献及Hale诊断标准,本文联合临床症状与基因检测对本院4例存在CHD7基因突变患儿进行诊断,其中3例可诊断为CHARGE综合征,患儿2虽有检测到CHD7基因可疑致病变异,但临床症状不明显,还不足以诊断为CHARGE综合征。4例患儿临床症状总结见表1。
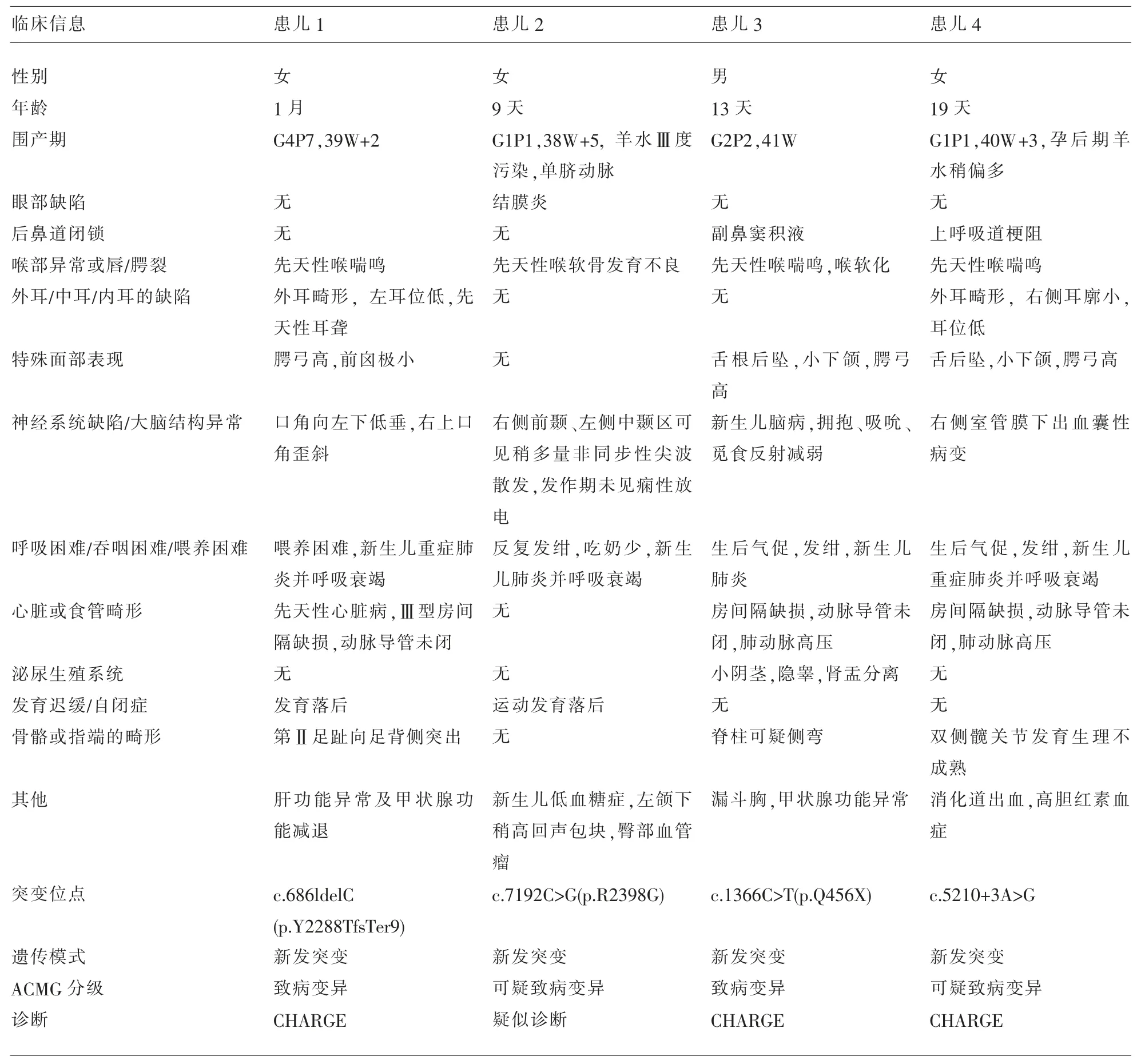
表1 4例携带CHD7基因突变患儿的临床特征
3 讨论
CHD7基因突变联合典型的临床表型可明确诊断CHARGE综合征[7]。CHD7广泛表达于胚胎和成体多种组织细胞中,不仅影响神经系统和生殖系统的发育,还广泛影响着眼、耳、心脏和骨骼等多个器官的发育,故CHARGE综合征患者的表型具有典型的异质性和复杂性[13],这种临床表现高度可变和缺乏强制性检查因素造成该病的诊断困难。最近,一种由全身系统和年龄组成的新检查表已被开发出来以帮助诊断CHARGE综合征[14],该检查表主要回顾了欧洲国家CHARGE综合征患者最常见的需要管理的问题,以及在这些患者的整个生命周期中医生可能忽略的关键问题,但目前还没有针对中国人群CHARGE综合征检查的全面指南。故本文报道4例存在CHD7基因突变患儿,总结其临床症状,为以后制定中国人群CHARGE综合征全面检查表提供一定帮助。
我们主要依据Hale诊断标准,4例患儿中有3例可以诊断为CHARGE综合征,其中患儿2现有的临床症状还无法诊断为CHARGE综合征,因该综合征表型的显著差异性,有些症状在新生儿期难以察觉,如视力、听力、性腺发育不良等[15],而该患儿亦未进行详细的全身检查,如眼球及视力检查、头颅CT,听力及半规管检查等,因此该患儿也无法排除CHARGE综合征,随后对患儿2进行的随访过程中,应注意对应CHARGE综合征常见表征进行随访。患儿1有明显的外耳畸形及听力异常、先天性心脏病、骨骼发育异常及宫外发育迟缓,加上CHD7基因(c.6861delC)致病突变可诊断为CHARGE综合征,c.6861delC为未报道过的新位点,该变异位点的检出扩展了CHD7基因的变异谱。据报道,80%~100%的CHARGE综合征患者存在耳朵异常,大多数外耳异常包括垂耳或杯状耳、耳廓低或垂直高度降低等[16-17],半规管发育不全是公认的CHARGE主要表征,可被认为是内耳畸形,在95%的CHARGE综合征患者中有听力损失表现,这些需通过CT或MRI检查来确认[18]。多数医院,包括我们医院,不使用CT或MRI作为初始筛查的一部分,除非有其他迹象证明需要[19],这可能导致一些表征难以发觉。在本研究中,患儿1和患儿4存在外耳畸形和耳位低,仅有患儿1表现先天性耳聋,其他2例患儿听力测试通过,这一比例低于文献报道比例,可能与本研究病例数较少有关。先天性心脏缺陷存在于3/4 CHARGE综合征患者中,其缺陷类型各不相同,从房室间隔缺损到主动脉弓畸形均有被报道[20]。本文报道的3例CHARGE综合征患儿均有先天性心脏缺陷存在,3例患儿缺陷类型均有房间隔缺损(ASD)和动脉导管未闭(PDA)。此外,在患者1还发生第2足趾突出,前囟极小症状;患者3出现肾盂分离及生殖器发育不良;患儿4有消化道出血、新生儿脑病和双侧髋关节发育不良,这些表征为CHARGE综合征少见的临床特征[21]。尽管CHARGE综合征患者的表型不同,但值得注意的是3例CHARGE综合征新生儿均出现先天性心脏缺陷、肺炎和先天性喉喘鸣,这表明当这些症状同时出现时,可被认为是CHARGE综合征的临床诊断线索之一。
本文总结4例携带CHD7基因杂合突变患者的临床表型,我们的研究表明,肺炎合并先天性喉喘鸣尤其是当它还伴有典型的结构畸形时可被视为CHARGE综合征的临床诊断线索之一,我们充分利用了基因检测的优势来诊断新生儿复杂的先天性疾病,这有助于医生做出针对性的治疗决策。本文报道的4例患者均为新生儿,CHARGE综合征的新生儿期诊断可提前对患儿多个器官系统进行评估与干预,尤其是神经系统和性腺轴中央激活情况,在随后的青春期进行早期干预、诱导,使其与同龄人同时发生第二性特征发育。基因检测可增进临床医生对CHARGE综合征发展和临床表现的了解,并可促进及时诊断,早期的支持和矫正疗法,特别是在控制感染,改善呼吸和进食方面,对患者的预后至关重要。由于基因检测的开展具有对CHARGE患者进行快速诊断和方便进行随访的好处,我们建议对不完全符合临床诊断标准的可疑病例进行基因检测。
