分子模拟在磷矿浮选研究中的应用进展
2023-01-17张覃李显波卯松章铁斌
张覃李显波卯松章铁斌
1.贵州科学院,贵州贵阳 550001;2.贵州大学矿业学院,贵州贵阳 550025;3.喀斯特地区优势矿产资源高效利用国家地方联合工程实验室,贵州贵阳 550025;4.贵州省非金属矿产资源综合利用重点实验室,贵州贵阳 550025
磷矿是我国战略性矿产资源,主要用于制备磷酸和磷肥等磷化工产品,是保障粮食安全和高新技术发展的物质基础,在国民经济中具有重要的地位和作用。据统计,截至2021年,世界磷矿石储量约为710 亿吨,主要分布在非洲、北美、亚洲、中东、南美等60个国家和地区[1]。按基础储量排名,摩洛哥和西撒哈拉位居第一位,中国位居第二位。我国对磷矿石的需求量逐年递增,2021年磷矿产量占世界总产量的39.06% ,居世界第一[1]。我国磷矿资源相对分布集中,中西部地区(云、贵、鄂、川、湘)占全国基础储量超过75% 。然而,我国磷矿资源禀赋差,存在“贫、细、杂”的特点。第一,磷矿石中P2O5的平均含量仅为17% ,P2O5含量小于30% 的中低品位磷矿储量为182.4 亿吨,占93% ,其中P2O5含量小于25% 的约占80% ,需要开展中低品位磷矿石选矿,来深度提磷、降镁、脱硅、脱倍半氧化物等;第二,磷矿石粒度细,呈均质胶体或隐晶、微晶质,集合体多为鲕粒、假鲕粒结构,鲕粒之间和内部常混入数量不等的碳酸盐、硅质等泥质矿物,需要开展微细粒磷矿石选矿;第三,磷矿床脉石矿物种类多、含量高,共伴生资源丰富,需要开展对磷矿中稀土、氟、碘、硅、镁、钾、铀、钛等共伴生资源综合利用。
沉积岩型磷块岩是世界各国磷矿的主要类型,我国磷矿储量中,87% 为沉积型磷块岩,分为钙(镁)质磷矿、硅质磷矿和硅钙质磷矿3 种类型。钙(镁)质磷矿中MgO 严重影响磷矿与硫酸反应,并降低磷酸使用和深加工的价值;As、Pb、Cd、Hg属于有毒有害元素,影响工业级和食品级磷酸质量,需要降镁、脱除重金属。硅质磷矿视SiO2、Al2O3和Fe2O3的形态不同,需要脱硅、脱除倍半氧化物(Al2O3和Fe2O3)。硅钙质磷矿中除SiO2和MgO 影响外,倍半氧化物易与磷酸反应生成多种磷酸盐,影响结晶特性,降低矿石P2O5利用率,需要脱硅降镁、脱除倍半氧化物。
先进的矿物表面测试技术可以对矿物表面微观区域进行细致的表征,但矿物浮选是发生在固-液-气三相表面的一个复杂的物理化学过程,研究矿物颗粒与颗粒之间、颗粒与水分子之间、颗粒与气泡之间、颗粒与药剂之间以及药剂与药剂之间的相互作用过程,采用这些测试技术不能完全解释在矿物表面发生微观变化的过程,同时也不能对这种复杂的物理化学过程进行准确的定义。如根据矿物表面测试的方法,能够明确药剂在矿物表面作用形成的产物,但是不能准确解释药剂作用的过程并对合适的药剂选择及设计进行预测及判断,而结合分子模拟,使得药剂的设计及选择更加精准,可以从矿物晶体结构、矿物晶格缺陷、矿物表面离子暴露机制、矿物表面能以及矿物表面与水分子及药剂分子作用的全过程准确地判断和预测,从而更加精准选择和靶向预测药剂分子结构,以及对矿物表面性质的变化进行解释和预测,为高效浮选药剂的选择作出精准判断。
本文重点回顾了磷矿浮选表面化学方法、分子动力学方法和量子化学方法,从分子模拟的角度总结了矿物晶体结构及表面性质、水分子和浮选药剂在矿物表面的相互作用,并讨论分子模拟在磷矿浮选研究中的应用前景。
1 磷矿浮选表面化学研究进展
基于矿物表面润湿性差异的泡沫浮选技术是实现磷矿石提质降杂的有效方法,世界上超过一半的磷精矿通过浮选法生产[2]。磷矿浮选表面化学是磷矿浮选的重要理论基础。20世纪80年代至90年代,主要开展了磷灰石、方解石界面行为和溶液化学研究,并在碱性正浮选条件下,发现矿物溶解离子导致矿物表面转化,矿物表面原子的价键特性是决定浮选剂选择性作用的关键因素,并开展了矿物颗粒表面间的界面极性相互作用研究。21世纪初,主要开展了浮选药剂及其浮选性能、细粒矿物聚集/分散行为与界面极性相互作用的关系研究,复杂离子对浮选影响以及白云石-胶磷矿体系中界面相互作用研究,从微观尺度研究了磷矿浮选表面润湿特性与吸附特性,以及微细粒分散絮凝行为。
Somasundaran 等[3]对浮选过程中盐类矿物(如磷灰石、方解石)的界面行为和溶液化学进行了系统研究。由于相似的物理性质和表面化学性质,分离碳酸盐矿物和磷酸盐矿物非常困难。碱性正浮选条件下,矿物溶解离子导致磷灰石和方解石表面发生转化。通过化学平衡计算,磷灰石和方解石在pH 值为9.8 时发生表面转化[4-5]。溶解离子导致的表面沉淀、溶解离子与表面活性剂生成沉淀,可能是碱性条件正浮选选择性分离丧失的原因[6]。
Liu 等[7]、Hu 等[8-9]等发现矿物表面原子的价键特性是决定浮选剂选择性作用的关键因素,揭示了矿物不同晶面及组分与浮选剂作用的各向异性、浮选剂亲固基团的取代效应,以及双极性基团强化与矿物作用的机制,设计了新型高效捕收剂和抑制剂,实现了浮选药剂与矿物之间的选择性作用,发现亲水矿粒表面水化力或疏水力主要归因于矿物颗粒表面间的界面极性相互作用[10]。Qiu 等[11]对白云石-胶磷矿体系中矿物界面相互作用进行系统研究,分析矿物晶体结构和矿物表面特性的差异,以及他们与矿物浮选行为之间的内在联系,从矿物晶体学、浮选表面化学的角度构建能够有效分离白云石与胶磷矿浮选的理论体系。Li 等[12]对石英表面特性与浮选的关系进行系统研究,从矿物晶体结构、浮选表面化学等揭示了石英表面特性与浮选的关系。
(1) 在润湿理论研究方面,俘泡法的测试环境与浮选体系相似,适合表征浮选体系中矿物颗粒的表面润湿性。Santos 等[13]采用俘泡法研究了荷荷芭油(Jojoba Oil)对磷灰石和方解石表面润湿性的影响。结果表明,pH 值为6.5 时,磷灰石完全亲水而方解石强疏水,两者能较好地实现反浮选分离。颗粒-气泡诱导时间能直观表征矿物颗粒与药剂作用后,颗粒与气泡的黏附作用以及表面的亲疏水性。采用诱导时间测定仪研究胶磷矿、石英和白云石与气泡、油泡和活性油质气泡间的作用,结果表明,脂肪酸活性油质气泡捕收矿物诱导时间最短,浮选回收率较高[14]。和频振动光谱(SFVG-S)是一种在分子水平研究表面或界面物理化学现象与过程的原位实时方法。Miller 等[15]研究了亲水和疏水二氧化硅表面水结构和润湿性,在亲水的二氧化硅与气泡接触过程中,其表面存在有序水结构的稳定水化膜。Lu 等[16]借助和频振动光谱分析了H2O2对石英界面水振动的影响,表明H2O2与石英表面的氢键比H2O 与石英表面的氢键更长。
(2) 在双电层理论研究方面,Zhou 等[17]采用Zeta 电位分布考察了活性油质气泡对磷灰石浮选的影响。结果表明,pH 值为9.01 时,活性油质气泡作用过的磷灰石能形成新的稳定表面,表明二者之间存在着比静电斥力更大的吸引力作用。添加Ca2+、Mg2+后,胶磷矿和白云石表面的电负性均降低,碱性介质下,PO34-对Ca2+、Mg2+在矿物表面上吸附作用影响较大,不利于矿物的浮选分离;CO23-和SiO23-在胶磷矿和白云石表面均有不同程度的吸附,但电负性差异并不明显。
(3) 在吸附理论研究方面,原子力显微镜(AFM)可获得原子间作用力分布信息,从而以纳米级分辨率获得表面形貌结构信息及表面粗糙度信息。Chennakesavulu 等[18]采用AFM 轻敲模式直观地观察了捕收剂在矿物表面的吸附,当捕收剂浓度低于1×10-7mol/L 时,捕收剂呈单分子层和双分层结构共存;当捕收剂浓度超过1×10-4mol/L 时,发生了多层吸附。Paiva 等[19]利用AFM 的接触模式直观地观察磷灰石表面形成吸附层和油酸钙的形貌,考察了pH 值对磷灰石表面形貌的影响。石英晶体微天平(QCM-D)是一种高灵敏度原位表面表征技术。Kou 等[20]采用QCM-D 考察了植物油和妥尔油脂肪酸捕收剂在磷灰石表面吸附,结果表明,QCM-D 可以很好地表征药剂在矿物表面吸附。Cao 等[21]采用XPS 对脂肪酸捕收剂在磷灰石表面的吸附进行研究,磷灰石表面Ca2p结合能改变,有羧酸钙等新物质生成,表面为化学吸附。此外,Wang 等[22]研究了C12TAB、SDS、SDDS、DAS 在石英表面的作用力,综合考察了临界胶束浓度(CMC)、表面张力、附着力等对药剂在石英表面吸附的影响。阳离子和阴离子表面活性剂在石英表面混合吸附,石英表面具有更强的疏水性。Yu 等[23]通过吸附量测定和吸附等温线研究表明,抑制剂NSFC通过化学键在白云石表面发生较强吸附作用,而通过氢键在磷灰石表面发生弱吸附作用,这种选择性吸附的差异导致其对白云石有较强的抑制作用,而对磷灰石抑制较弱。随着酸用量增加,GJBW 在白云石表面吸附量逐渐增加,而在胶磷矿表面吸附量降低,二者吸附量差异是导致其浮选回收率差异的主要因素,GJBW 在白云石表面以化学键作用为主[24]。胶磷矿自身溶出的H2PO-4 是主要的抑制组分。溶出离子种类和浓度取决于初始氢离子浓度和反应后矿浆pH 值。酸浓度对捕收剂吸附量影响较小,但能强烈抑制胶磷矿上浮;H2PO-4 与捕收剂分子在矿物表面不是竞争吸附,而是共同吸附,H2PO-4 在胶磷矿表面占主导地位,导致表面亲水[25]。
2 分子模拟在浮选中研究进展
随着理论化学和计算化学的快速发展,分子模拟已成为研究磷矿浮选的有效手段,为分子或原子水平研究矿物晶体结构、表面性质和界面相互作用提供新的思路[26]。分子模拟计算能从Mulliken 电荷布局、Mulliken 键布局、吸附能、态密度、电荷密度等方面研究表面润湿性、表面电性和表面吸附。
陈建华[27]系统研究了硫化矿物晶体电子结构、表面结构和电子性质,矿物表面与浮选药剂分子相互作用以及晶格缺陷对硫化矿物半导体性质及可浮性的影响。Gao 等[28]利用分子模拟研究了十二胺在白钨矿和方解石表面上的吸附,结果表明,十二胺在白钨矿和方解石表面的不同吸附行为主要归因于十二胺中的阳离子RNH3+。Wang等[29]通过分子动力学模拟单磺酸盐与双磺酸盐对萤石浮选的差异,结果发现,磺酸活性物质的分子量起到抑制作用,浓度较大的双磺酸盐会产生空间位阻效应。Zhang 等[30]通过分子动力学模拟发现,Ca-蒙脱石中腐殖酸、羧基和钙离子之间的络合反应在腐殖酸的吸附过程中占据主导作用。Han 等[31]采用前线分子轨道理论分析发现,黄原酸异丁酯比黄原酸丁酯更容易吸附在黄铁矿表面。Chen 等[32]研究了海藻酸钠在不同捕收剂体系下方铅矿浮选的抑制作用,分子模拟结果表明,丁基黄药对方铅矿的吸附能力大于海藻酸钠。Chen等[33]通过DFT 研究了典型杂质对闪锌矿电子结构的影响,结果表明,镉、汞、砷等杂质可以增大闪锌矿的晶格参数,银、锡、铅等杂质提高了闪锌矿的导电性。
3 分子模拟研究方法
3.1 分子动力学方法
分子动力学模拟(Molecular Dynamics Simulation,MDS)多使用经典分子动力学方法[34-35]。通过求解牛顿运动方程,描绘粒子的运动轨迹及其随时间变化的过程,结合统计力学确定体系的宏观性质[36]。经典分子动力学计算采用简化的原子模型,不考虑电子云结构,将原子核及核外电子看作一个粒子,计算时仅考虑由粒子间相互作用引起的能量变化,该方法可在三维空间填充大面积的晶体表面、液体团簇或气体团簇,采用经典运动方程与原子间力的迭代求解可模拟出体系结构和能量的变化过程[37]。浮选领域研究者使用MD 方法研究矿物表面水化层、药剂在矿物表面的吸附构型和吸附行为、颗粒-颗粒相互作用和颗粒-气泡相互作用[38],为了解矿物表面性质、高效筛选浮选药剂提供理论指导。
MDS 基于分子力学(Molecular Mechanics,MM),采用经典力学方程求解核运动,采用经验势函数表征模拟对象间的相互作用。分子力学用于计算分子间相互作用的函数和参数集称为力场,力场的基本函数表达式[39]如下:
Etotal=Ebonded+Enon-bonded
Ebonded=Ebond+Eangle+Edihedral+Eout-of-plane
Enon-bonded=EVanderWaals+Eelectrostatic+EH-bond
式中,Etotal为分子力学计算需要求解体系分子间的相互作用势;Ebonded为分子内成键作用势;Enon-bonded为分子间相互作用势;Ebond为键伸缩势;Eangle为键角弯曲势;Edihedral为二面角扭转势;Eout-of-plane为离平面弯曲势;EVanderWaals为Van der Waals 势;Eelectrostatic为静电相互作用势;EH-bond为氢键势。
分子力学计算的准确性主要取决于力场,但力场是经验性的,没有完全适用于所有体系的力场。常见的力场有OPLS 力场、AMER 力场、CHARMMI力场等[40]。随着技术的发展,为特定体系构建的特殊力场如PCFF 力场[41]和CLAYFF 力场[42]也在逐渐完善,被广泛应用于研究矿物-水分子、矿物-药剂的相互作用。
3.2 量子化学方法
量子化学计算是基于量子力学的理论和方法,假设原子之间的相互作用都与电子有关,通过求解薛定谔方程描述粒子的运动。但多粒子系统相互作用的复杂性导致求解薛定谔方程十分困难,常采用多种近似方法来简化计算过程。其中,密度泛函理论(Density Functional Theory,DFT)改变传统以轨道波函数为基础的特点,用粒子密度函数表述系统基态各物理量,以电子密度函数来表示体系能量[43]。
采用DFT 模拟磷矿中氟磷灰石和白云石等矿物的浮选行为,一般按照以下步骤进行:
(1) 构建合理的矿物模型,包括氟磷灰石和白云石等矿物。
(2) 对构建的矿物模型进行几何优化,获得结构稳定的构型。
(3) 以稳定矿物模型为对象,以XRD 衍射峰对应的晶面为依据,构建矿物表面,得到与实际矿物解理面一致的矿物表面。
(4) 扩展表面结构,为药剂吸附预留足够的空间,即构建一定厚度的真空层结构。
(5) 在选定表面上适当位置放入药剂分子,模拟吸附过程,通过不断调整药剂在表面的吸附位置,并对比吸附能,最终确定药剂在矿物表面的吸附构型。
MDS 和DFT 是目前广泛使用的分子模拟方法,在精度、模拟对象、计算成本、系统大小和模拟时间方面各有优势。DFT 能精确描述涉及电子转移并形成共价/配位键的相互作用,可以对矿物内部结构、表面电子性质、药剂和矿物表面的成键作用等进行精确研究;不足之处在于复杂反应体系的模拟对计算硬件资源要求高,耗费机时较多,导致一些复杂的浮选作用过程模拟难以完成。基于牛顿运动定律的MDS,是计算复杂分子体系的有效方法,其可以模拟数千个原子至百万个原子组成的体系,已成为一种从原子尺度研究表面活性剂在界面上的自组装行为、界面水结构以及矿物表面润湿性的有效手段[44-45]。但MDS 主要考察分子间作用力且模拟过程中必须输入力场模型,多数经典力场对远离平衡位置的化学反应无法描述。
通过对矿物表面与药剂相互作用的分子模拟计算,能够在分子水平提供药剂与矿物相互作用的复杂模型,加深对药剂与矿物之间相互作用的理解,为实际矿石的浮选提供理论依据[46]。分子模拟方法受计算模型和硬件条件的限制,研究结果尚存在一定局限性,在运用分子模拟方法开展选矿研究时,要将其与试验结果相印证;同时将MDS 与DFT 有机结合,从而实现电子尺度的量子化学分析和复杂体系原子尺度的动力学研究有机结合。
4 分子模拟在磷矿晶体化学和界面相互作用研究中的应用
在浮选过程中,矿物晶体结构、矿物表面特性、浮选药剂及其相互作用关系是影响浮选效果的内在关键因素。计算化学的发展成熟和计算机硬件条件的提升,使得通过分子模拟计算对矿物的结构、表面以及药剂作用的研究成为可能。针对浮选实际情况,采用分子模拟计算对矿物内部结构、表面微观性质、浮选药剂分子进行分析,解释选矿过程中所表现出来的宏观现象,并为浮选药剂的合成与开发提供指导[47-48]。图1所示的Web of Science 检索结果表明,自2004年以来,以 “flotation” and “molecular simulation” 和“flotation” and “DFT” 为关键词统计的年均发表相关论文数量呈不断增加的趋势,特别是近10年来,分子模拟方法在浮选过程作用机制的基础研究中,得到越来越多的关注。

图1 以“flotation”and “molecular simulation” 和“flotation” and “DFT” 为关键词的年均发表相关论文数量Fig.1 Annual average papers published with the keywords “flotation” and “molecular simulation”,“flotation” and “DFT”
4.1 矿物晶体结构及表面性质的DFT 研究
矿物的可浮性由其晶体结构和化学组成共同决定,结合分子模拟对不同矿物晶体建模和处理,实现对矿物晶体化学性质、矿物表面性质的研究,为高效分选磷灰石提供理论指导。理想氟磷灰石晶格常数为a=b=9.375 Å,c=6.887 Å,α=β=90°,γ=120°,空间群为C26h-P63/m,晶胞中两组Ca1 原子(每组2个)形成垂直(001)面的原子柱,两组Ca2 原子(每组3个)形成垂直于c轴的端面[49]。如图2所示,氟磷灰石晶体中存在Ca1、Ca2 两种位点,Ca1 位点处于两层磷酸根之间,Ca2 位点与磷酸根处于同一层[50]。对两种位点的成键特性进行研究发现,Ca1 原子与周围9个氧原子成键,有效配位数7.456;Ca2 原子与临近6个氧原子和F 原子成键,有效配位数6.318[51]。氟磷灰石中Ca 与临近原子的成键过程中,3个O3 的2s 轨道与Ca1 的3p 轨道形成3个弱σ 键,3个O3 的2p 轨道与Ca1 的3d 轨道成弱π 键,成键中离子键成分更大[52]。由于成键(σ 键或π 键)与反键(σ*键或π*键)间劈裂程度大,对应O—P 键强较大,对键强贡献较大的为σ 键和σ*键,O—P 之间主要形成共价键(Mulliken 键布居值约0.6),因此该键一般不易断裂。Ca2 原子成键的Mulliken 键布居值大于Ca1 位点成键,导致Ca1 位点活性较强。1个Ca2 的3p 与F 的2s 形成σ 键,同时1个3d 与F2p成弱π 键,F—Ca2 键总体呈离子性[52]。对氟磷灰石常见解理面(001)的三种端面(Ca1、PO4和Ca1—Ca1)的表面能计算结果表明,Ca 端面、Ca—Ca 端面和O 端面的表面能分别为0.811 J/m2、1.541 J/m2和1.425 J/m2,Ca1 端面表面能最低,进一步证明了Ca1 位点处由于活性更强,易产生不饱和断裂键[53]。
高超走上台阶按响了门铃,他们等了两分钟,开门的还是罗云。高超出示了证件和搜查证,罗云闪身做了个请他们进屋的动作,随手关上门,把寒风挡在门外。看见老福她依然没开口,好像从来没见过他一样。
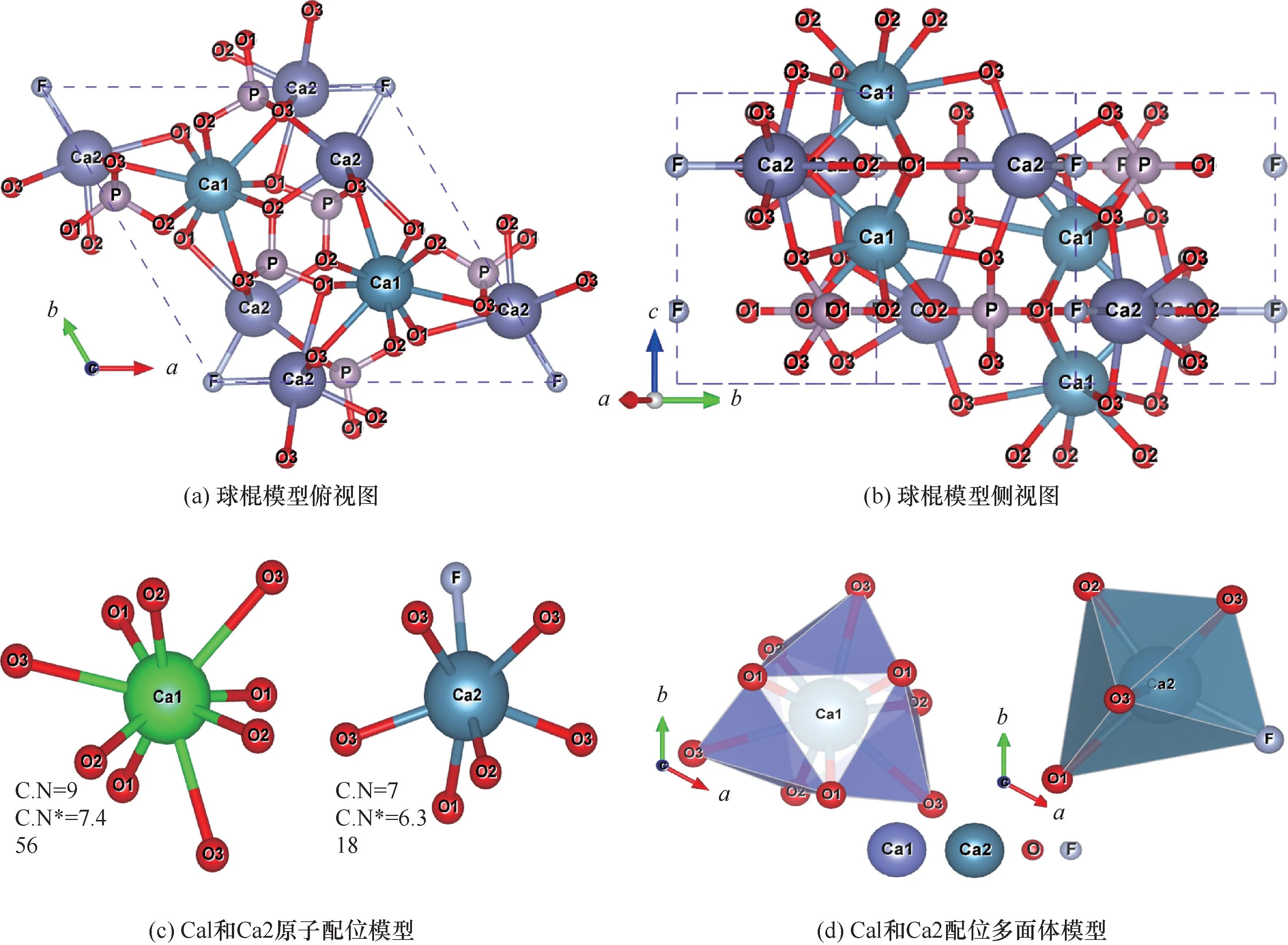
图2 氟磷灰石晶体模型及Ca1 和Ca2 配位模型[49]Fig.2 Crystal model of fluorapatite and coordination models of Ca1 and Ca2 [49]
对氟磷灰石的晶面各向异性分析发现,氟磷灰石常见暴露面的表面能和断裂键密度满足(001)>(100)>(101)>(111),且4个表面的粗糙度和润湿性也满足(001)>(100)>(101)>(111),表明单位面积断键数是影响各向异性润湿特性的主要因素[54]。在晶体生长过程中,(001)晶面方向逐渐增大,(101)表面逐渐减小,理想氟磷灰石晶体的双锥最终消失,并形成柱状。氟磷灰石实际晶体可能存在于如图3所示的任何形态中。无论氟磷灰石晶体的形态如何,都可能出现(100)、(001)、(101)和(111)表面。成矿过程中由于环境的影响,实际矿物晶体通常存在各种晶体缺陷,通过对理想晶体中的特定原子进行删除、移动或替换可以模拟矿物的晶体缺陷,从而研究晶格缺陷对矿物性质的影响[55]。

图3 氟磷灰石在真空条件下生长的晶体形态[54]Fig.3 Crystal morphologies of FAP bulk grow in vacuum [54]
针对含稀土磷矿石,谢俊等[56]认为稀土元素以类质同象形式赋存于磷灰石中。稀土原子在氟磷灰石晶体Ca 位点掺杂的DFT 计算表明,稀土原子在Ca 位点取代能呈“斜W 形”且具有明显的“钆(Gd)断现象”,稀土原子在Ca1 和Ca2 位点取代能的变化趋势基本一致,取代能总体上呈负值,理论上稀土原子均可在Ca1(或Ca2)位点发生取代,稀土在Ca 位点取代能如图4所示。重稀土倾向占据Ca1 位点,轻稀土倾向占据Ca2 位点,取代作用以空间调节机制为主[49,57]。

图4 稀土在不同Ca 位点的取代能[49,57]Fig.4 Substitution energies of rare earth atoms at different Ca sites[49,57]
4.2 水分子与矿物表面相互作用的DFT 和MDS研究
浮选过程中,矿物表面暴露在水溶液环境中,形成水化层,不利于捕收剂的吸附[58]。研究矿物表面与水的相互作用有助于深入理解矿物表面性质。随着计算科学和技术的发展,结合DFT 方法和MDS 方法研究矿物表面水化作用已成为常用手段。
使用DFT 研究单个水分子在氟磷灰石(001)面的吸附(图5)发现,氟磷灰石白云石、方解石和表面具有不饱和的Ca/Mg 金属活性位点,水分子在其表面均可以发生强烈吸附,水分子中的OW原子与表面Ca 原子形成牢固的Ca—OW键,水分子中的HW则与矿物表面的OS原子形成氢键[50];水分子在羟基磷灰石和(001)面的吸附也呈类似规律[59]。
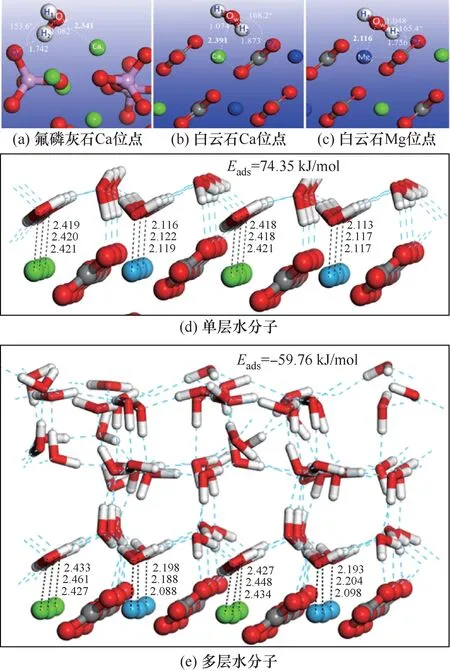
图5 单个水分子、单层水分子和多层水分子在氟磷灰石和白云石表面吸附[60]Fig.5 Adsorption of single,single-layer and multi-layer water molecules on fluorapatite and dolomite surface[60]
水分子在白云石(104)表面的吸附主要形成Ca/Mg—OW键,在白云石表面Mg 位点吸附过程中,主要是Mg3s、Mg2p 与O2s、O2p 的成键和反键作用,且Mg 位点的吸附作用弱于Ca 位点,这种差异可能是由于两种原子的外层电子数和原子半径导致的[60]。相关研究认为,Ca、Mg 原子半径不同,且在白云石表面交替排列,因此矿物表面CO23-呈一定偏转角排列[61];水分子吸附在Ca 位点时形成的氢键比吸附在Mg 位点短[62]。
单层水分子在白云石表面的吸附模拟结果显示,吸附在Ca 位点的水分子倾斜排列,吸附在Mg位点的水分子呈“V”形排列[63];原子力显微镜成像进一步证实了水分子在Ca、Mg 位点的吸附差异,与Ca 位点相比,Mg 位点上吸附的水分子距离表面更近[64]。
经MDS 模拟发现,水滴可在氟磷灰石、方解石和白云石表面有效铺展,即水滴簇可以很好地在3种矿物表面润湿(图6)。这与磷灰石、方解石和白云石自然表面亲水的认知是一致的。水分子簇在白云石表面吸附能(-8.13×103kJ/mol)略大于方解石表面(-7.51×103kJ/mol),但二者均比水分子簇在氟磷灰石表面吸附能大(-4.78×103kJ/mol)[60]。

图6 水滴在氟磷灰石、方解石和白云石表面润湿动力学[60]Fig.6 Wetting kinetics of a water droplet on apatite,calcite and dolomite surfaces [60]
磷酸盐和碳酸盐矿物在表面生成过程中产生较多的不饱和悬挂键,表面有较强的极性,对极性水分子具有较强的吸引力,表面表现出较强的亲水性。Pareek 等[67]采用掠入射X 射线衍射研究了水在氟磷灰石(100)表面弛豫中的作用,发现在矿物表面1.6 Å 和3.18 Å 处分别形成两层致密水结构。利用DFT 和MDS 还可对羟基磷灰石与表面水化层结构的相互作用进行探索。潘海华等[66]采用MDS 对羟基磷灰石(100)和(001)晶面上的水化研究表明,在羟基磷灰石表面可形成2 ~3 层有序的水分子层,表现出类似冰的致密结构,且(001)方向的极性更强,可以形成更多有序结构化水层。
4.3 浮选药剂与矿物表面相互作用的DFT 和MDS 研究
磷矿浮选中常采用脂肪酸类捕收剂,主要包括脂肪酸及其盐类、塔尔油和氧化石蜡皂[68-69]。脂肪酸具有较好的捕收和起泡能力,且成本较低,油酸及其油酸盐对磷灰石的捕收作用机理已被广泛研究[70-71]。脂肪酸类捕收剂通常与矿物表面金属离子相互作用,使矿物疏水[72]。对氟磷灰石(001)面的电子性质研究发现,氟磷灰石表面Ca、O 原子活性较强;与体相中的Ca 原子相比,表面Ca 原子的电子态密度向费米能级方向移动,表面Ca 原子荷电1.37个电子,使氟磷灰石表面呈金属性,容易与阴离子发生相互作用[44]。
基于DFT 理论研究脂肪酸类捕收剂阴离子在氟磷灰石表面的吸附(图7)发现,脂肪酸捕收剂的O 原子与氟磷灰石表面的Ca 原子形成化学吸附,脂肪酸的H 原子与氟磷灰石表面中的O 原子形成氢键吸附,脂肪酸阴离子在氟磷灰石表面的吸附主要取决于表面Ca 原子和阴离子中O 原子之间的相互作用[53]。采用DFT 模拟研究脂肪酸类捕收剂的碳链长度、碳链异构和C==C 双键数对药剂在氟磷灰石(001)面的吸附影响。结果表明,随着碳链在一定范围内增长,捕收剂O 原子与表面Ca 原子态密度曲线重叠性增加,吸附作用增强;C ==C 双键数对药剂的吸附作用无明显影响;碳链异构对脂肪酸在氟磷灰石表面的吸附影响较大,对比4-甲基庚酸和6 -甲基庚酸在Ca 位点的吸附结果,4-甲基庚酸的吸附模型中Ca—O 间电荷密度更大、Ca 原子和O 原子态密度曲线重叠性增加、吸附能绝对值增加[53]。在碱性条件下,Mg2+和Al3+离子在氟磷灰石表面形成MgCO3和Al(OH)3沉淀后,油酸阴离子能够与氟磷灰石表面形成更强的Al/Mg—O 和Ca—O 的双键吸附[73]。

图7 乙酸、戊酸和辛酸在氟磷灰石表面的吸附构型[53]Fig.7 Adsorption configuration of acetic acid,valeric acid and caprylic acid on the fluorapatite surface[53]
白云石和氟磷灰石在自然pH 值条件下可浮性相似,难以高效分离,通常在酸性条件下使用硫酸和磷酸作为调整剂抑制磷灰石上浮,使用油酸类捕收剂反浮选白云石。研究认为,酸根离子与磷灰石表面Ca2+形成难溶性盐沉淀覆盖于矿物表面,降低了捕收剂吸附量[74-75]。采用DFT 模拟研究硫酸根和油酸阴离子在白云石表面吸附构型发现,白云石在酸性条件下形成CO23-空位缺陷后,SO24-能够与缺陷处的Ca2 结合,但是油酸阴离子仍能吸附在剩余的Ca 位点和Mg 位点处,导致白云石疏水[76];研究溶液中游离的Ca2+对矿物表面性质的影响发现,Ca2+位于氟磷灰石和白云石表面时,油酸阴离子与氟磷灰石的相互作用减弱,油酸阴离子的羧基中两个O 原子与Ca2+形成双键,Ca2+增强了油酸阴离子在白云石表面的吸附[77]。如图8所示,无捕收剂时,水滴可以在氟磷灰石表面完全铺展;油酸钠(NaOL)极性基可克服表面水化层作用而与矿物表面金属位点形成稳定吸附,这是实现矿物表面疏水性调控的前提。在氟磷灰石(001)表面,NaOL 单分子层吸附结构可以形成良好的疏水表面,而双分子层吸附则导致表面亲水,水分子与NaOL 极性基的氢键缔合作用是双层吸附表面亲水的本质。

图8 水分子团簇在不同NaOL 吸附层结构的磷灰石(001)面润湿性接触角MDS 构型[60]Fig.8 MDS results of contact angle of water droplet on apatite(001)surfaces with different structure of sodium oleate adsorption layer [60]
仪器分析受限于样品中待测元素含量和仪器的检测下限,相比较而言,采用分子模拟方法可以实现常规条件下难以进行的研究,如基于DFT 计算,可以从能量、成键、态密度、电荷密度、Mulliken布居等角度解释轻/重稀土取代氟磷灰石Ca 位点的难易程度,水分子和药剂分子在氟磷灰石表面的吸附位点、成键特性及电子转移情况,明确捕收剂分子结构对脂肪酸在氟磷灰石表面的吸附影响;基于MDS 计算,可以直观观察到水滴在矿物表面形成的水化层结构,揭示矿物表面自然亲水和脂肪酸捕收剂单分子层吸附形成疏水表面的微观本质,使磷矿浮选机理研究深入到分子/原子水平。
5 展 望
随着表面测试手段的进步,可以采用接触角测定仪、诱导时间和SFVC 研究表面润湿性,采用Zeta 电位及纳米粒径分析仪研究表面电性,采用AFM、QCM-D、XPS、Micro Calorimeter、FBRM、TOF-SIMS 研究表面吸附。采用分子模拟可以计算Mulliken 电荷布居、Mulliken 键布居、吸附能、态密度、电荷密度等。新的原位、直观、高精度的微观表面测试手段不断在浮选研究中得到应用的同时,加强DFT 和MDS 分子模拟计算在浮选研究中的应用,可以不断丰富和发展浮选表面化学理论。
矿物的可浮性由其晶体结构和化学组成共同决定,采用分子模拟研究氟磷灰石、白云石和方解石的晶体化学性质以及水分子与浮选药剂在矿物表面的吸附,能够为磷矿石的高效分选提供一定的理论指导。单独使用MDS 和DFT 均存在一定的不足,对部分体系仍不能较好描述。随着计算能力的提高,将分子力学与量子力学方法有机结合,开发出基于量子力学理论的第一性原理分子动力学(Ab Initio Molecular Dynamics,AIMD)模拟,通过量子化学方法直接计算所有分子内和分子间相互作用,不用输入经验的力场,为研究矿物的晶体和表面结构、表面水化和润湿性、浮选药剂的吸附构型和相互作用行为,以及颗粒-颗粒或颗粒-气泡的相互作用提供有益的技术支撑。
随着分子模拟的快速发展,必将促进浮选药剂与矿物表面作用的研究,将为高选择性药剂的研发提供新的原子层面的分析技术,这对开发利用日益复杂的矿产资源有重要的理论和实际意义。磷矿浮选研究深入到分子/原子水平,能获得更多精确判断化合物性质的量子化学参数,以此进一步定量解释矿物表面电子性质以及捕收剂的构效关系。在氟磷灰石、白云石和方解石三者浮选分离调控研究中,需要针对其阴离子差异或表面活性位点分布特性来匹配合适的抑制剂。通过研究氟磷灰石、白云石、方解石表面活性位点的分布机制,有利于靶向设计钙镁质磷矿石高选择性捕收剂和抑制剂。
