埃希氏菌的遗传演化关系分析
2023-01-16刘俊华周忆莲崔垚鑫刘燕霏杨建德
刘俊华,周忆莲,崔垚鑫,刘燕霏,杨建德
(天津农学院 动物科学与动物医学学院,天津市农业动物繁育与健康养殖重点实验室,天津 300392)
埃希氏菌(Escherichia)是重要的人畜共患病病原菌,目前已知有七个物种,分别是大肠埃希氏菌(CASTELLANI&CHALMERS,1919)、赫尔曼埃希氏菌、伤口埃希氏菌(BRENNER等,1982)、弗格森埃希氏菌(FARMER等,1985)、蟑螂埃希氏菌、艾伯特埃希氏菌(HUYS等,2003)以及从中国青藏高原旱獭粪便中发现和命名的旱獭埃希氏菌[1-3]。大肠埃希氏菌俗称大肠杆菌,有一部分血清型常引起幼龄动物发生严重腹泻和败血症等疾病,且这类具致病性的大肠杆菌能在不同种动物之间进行传播[4-6]。赫尔曼埃希氏菌是条件性致病菌,可引起人的原发和继发性感染,如呼吸道、泌尿系统感染及伤口化脓性感染等[7]。弗格森埃希氏菌是对人类少见的条件致病菌,可引起人和动物创伤感染、尿道感染、菌血症、腹泻和胸膜感染等[8-11]。艾伯特埃希氏菌在一处人类病例中导致患病者出现血性/非血性腹泻、 呕吐、脱水、发热、腹胀等临床症状,是一种重要的潜在新发食源性致病菌[12]。蟑螂埃希氏菌、旱獭埃希氏菌经查阅现有资料未得到具体临床症状信息。埃希氏菌在致病性上或多或少诱发人畜表现出相同或相似的临床症状,同时又致人畜表现出其他完全不同的临床症状,使疾病防治难度加大。
16S rRNA是所有原核生物蛋白质合成必需的1种核糖体RNA,大约由1 550个核苷酸组成,其长度能够表现足够的种间多态性,又便于序列分析[12]。细菌基因组中16S rRNA在漫长进化中表现出高度保守性,有微生物“化石”之称。它的结构中保守区域与变异区域共存,形成了不同微生物的特征核苷酸序列,是较好的生物标志物,可作为微生物多样性分析的分子基础,用来鉴定细菌的进化距离和亲缘关系。2005年,基于焦磷酸测序法的高通量测序系统为测序技术带来革命性进步,16S rRNA数据库得到迅速扩大[11]。本试验通过对实验室分离菌及 NCBI数据库检索的多种埃希氏菌菌株的16S rRNA进行筛选、整理,经同源序列比对分析构建分子进化树,探究不同地区埃希氏菌的进化与遗传演化关系,揭示埃希氏菌在畜禽疾病传播与突变中的遗传进化规律,为防治该病提供理论参考依据。
1 材料
在本实验室培养并分离出4株菌株,GenBank序列号分别为MK729003.1(E. coli1 106,1 481 bp)、MK729001.1(E. fergusoniijoe,1 483 bp)、MK729002.1(E. coli1 018,1 475 bp)、MK685226.1(E. marmotaeEy-zq,1 480 bp)。其余16S rRNA序列均来自美国国立生物技术信息中心(The National Center for Biotechnology Information,NCBI),进入NCBI主页(https://www.ncbi.nlm.nih.gov)选择需要的序列进行下载。
BioEditv7.0.5软件下载自 http://www.mbio.ncsu.edu/BioEdit/bioedit.html,MEGA X软件下载自https://www.megasoftware.net,DNASTAR软件下载自https://www.dnastar.com。
2 方法
2.1 基因序列筛选
在 NCBI网站搜索框中,通过 Nucleotide BLAST将实验室分离的 4组基因序列分别运行BLAST比对,检索NCBI数据库中16S rRNA序列。本试验以埃希氏菌属16S rRNA作为研究范围,探究不同地区埃希氏菌的进化与遗传演化关系。试验排除了序列检索到的非埃希氏菌基因序列信息,而选择BLAST比对结果中同源相似性较高、序列一致性在95%以上的序列[12-13]。同时检索其他未进行比对的埃希氏菌16S rRNA序列,以此扩大试验菌株的地域范围及菌株多样性。对这些序列进行下载,在 NCBI中收集序列信息,包括 GenBank序列号、菌株编号、序列来源地区、序列长度,整理数据。
2.2 基因同源性比较及分子进化树构建
2.2.1 使用BioEdit软件对基因序列进行处理
打开 BioEdit软件运用 ClustalW Multiple alignment方法对42个基因序列进行整合、比对、分析,得到长度约为1 100~1 483 bp之间的基因序列。
2.2.2 基因同源性比较
打开 DNASTAR软件导入已做处理序列,进行基因同源性比较。
2.2.3 利用MEGA X软件构建分子进化树
在 MEGA软件中,用 Neighbor-Joining(NJ,邻接法)和 p-distance模型按照软件原始参数构建分子进化树使已经修剪对齐的 16S rRNA基因序列生成邻接树,估计它可以引导支持运行100 000次重复检测[13-14]。同时采用Bootstrap法进行Bootstrap values(步长值)为1 000的重排构树检验所计算的进化树各分支的可信度,估计 95%的可信限度,完成分子进化树构建[15]。
3 结果与分析
3.1 基因筛选
基于16S rDNA序列(16S rRNA基因)分析是细菌系统分类的“金标准”[16],在NCBI数据库中分别以 MK729003.1、MK729001.1、MK729002.1、MK68526作为目的基因进行搜索,通过BLAST中的Nucleotide BLAST选项,共得到400个部分重复的基因序列,同时在NCBI总数据库中搜索得到100个埃希氏菌16S rRNA基因序列,经过进一步筛选,得到42个来源明确、长度适中的埃希氏菌16S rRNA基因序列,见表1。
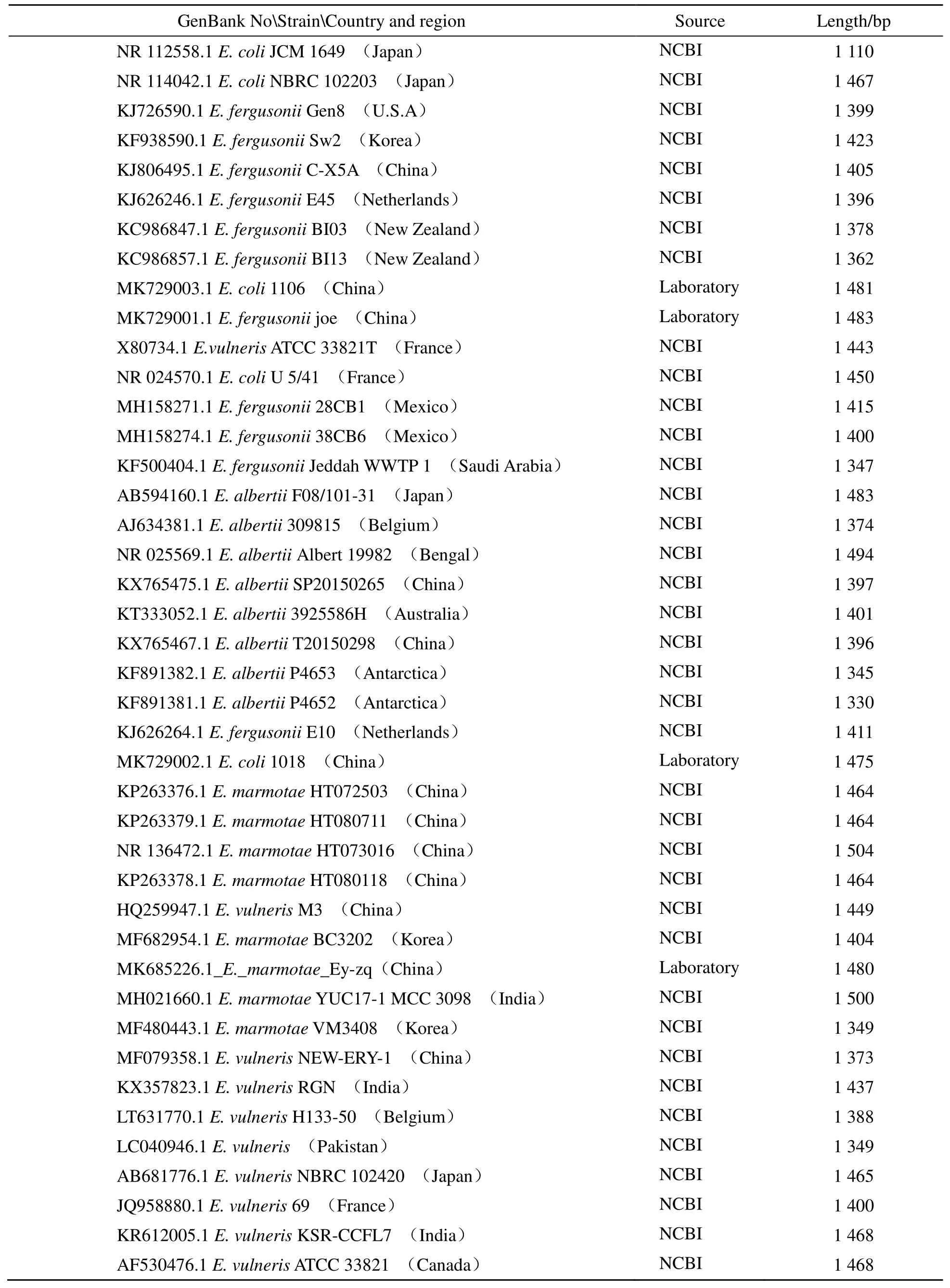
表1 埃希氏菌属16S rRNA基因序列信息
3.2 基因同源性比较及分子进化树构建
根据表1数据,运用DNASTAR、MEGA X软件进行同源性比较和序列比对,用邻接法构建邻接分子进化树,见图1。

图1 42株埃希氏菌株分子进化树分析
由图1可以看出,所有菌株根据在进化树中的分布可以分为四大分支:大肠埃希氏菌与弗格森埃希氏菌集中分布一支,艾伯特埃希氏菌、旱獭埃希氏菌及伤口埃希氏菌分成三支且各自独立集中分布。四大分支遗传距离远近呈现有序的状态,总体来看,旱獭埃希氏菌分支在进化树中结点最为靠前,其后依次是大肠埃希氏菌、弗格森埃希氏菌、艾伯特埃希氏菌、伤口埃希氏菌。伤口埃希氏菌绝大多数与其他四种菌处于完全不同的一支上,同源性仅在 94.0%左右。同时在大肠埃希氏菌、弗格森埃希氏菌、艾伯特埃希氏菌、旱獭埃希氏菌中,或多或少的有某些菌株交叉在一起,同源性在 96.0%左右,如 HQ259947.1E.vulnerisM3 (China)远离本菌群分支靠近旱獭埃希氏菌分支,与MF682954.1(Korea)同源性98.8%,与MK685226.1(China)同源性99.6%,另外大肠埃希氏菌与弗格森埃希氏菌完全混杂在一起,大部分菌株同源性高达 98.0%。这反映出四大分支菌的基因同源性大小和亲缘关系的远近,也反映出菌株之间存在着程度不同变异,且不同类种菌株间相互变异度不同,推测大肠埃希氏菌和弗格森埃希氏菌之间变异度最高,其余分支在菌种之间变异较低,存在较高保守性[16-18]。
在进化树中,每一大分支中多数菌株支长存在较小差异,且有着非常明显的地域特征。这些基因序列在较小的距离内发散,最终聚类到一起,且各类聚合支之间的地域特征非常明显[19]。
3.2.1 大肠埃希氏菌和弗格森埃希氏菌共同进化
大肠埃希氏菌和弗格森埃希氏菌混杂在一起,它们的遗传距离较近,如 NR 114042.1E. coliNBRC 102203 (Japan)与 KJ726590.1E. fergusoniiGen8 (U.S.A)的同源性高达98.9%,MK729003.1E. coli1106 (China)与 KC986847.1E. fergusoniiBI03 (New Zealand)、KC986857.1E. fergusoniiBI13 (New Zealand)、MK729001.1E. fergusoniijoe(China)的同源性分别为99.3%、99.1%、100.0%。另外,KJ626264.1E. fergusoniiE10 (Netherlands)与MK729002.1E. coli1018(China)分支相距较远,但这两种不同菌种菌株的同源性高达 99.4%。由此推测两种菌在很早的时期是由共同的原始菌株演化而来,且两种菌之间发生变异而相互转化的可能性也极高[19]。在图 1 中,KF938590.1(Korea)、KJ806495.1(China)与 KJ626246.1(Netherlands)聚为一支,KC986847.1(New Zealand)与KC986857.1(New Zealand)、MK729001.1(China)、MK729003.1(China)聚为一支,这两支同源性高、遗传距离较近,且前一支结点靠前,推测弗格森埃希氏菌可能在跨区域传播过程中发生变异形成新的本菌群菌株,部分菌株变异成大肠埃希氏菌属[17-18]。
3.2.2 艾伯特埃希氏菌单独进化
由图1也可以看出在艾伯特埃希氏菌中,来自南极的KF891382.1(Antarctica)和KF891381.1(Antarctica)两株聚类到一支,同源性高达98.2%;来自中国的KX765467.1 (China)远离KX765475.1(China)且与之同源性高达 99.8%,两株菌株都远离来自日本的AB594160.1 (Japan),却与南极的两株聚类到一个结点,同源性分别为 97.9%、96.8%、97.7%、96.6%;同时KX765475.1(China)与 KT333052.1(Australia)聚类为一支,同源性高达 99.2%。由此可以推测,该菌型在同一地区存在高度保守菌株与保守性较低菌株共存的情况,且在亚洲与大洋洲、南极洲三大洲间存在着交叉进化。
3.2.3 旱獭埃希氏菌两枝进化,与伤口埃希氏菌关系较为密切
在图1中,可以发现一个明显的中国地区旱獭埃希氏菌聚集,KP263376.1 (China)、KP263379.1(China)、NR 136472.1(China)、KP263378.1(China)四株构成了进化较近的中国基因型,呈现一种独立的进化分支,并远离其他分支,说明旱獭埃希氏菌在我国存在特有的菌型,其具有高度的保守性,但从图中仍能够发现另一株MK68526(China)不在本分支内,体现出中国旱獭埃希氏菌分支的存在。从图中还能发现MF682954.1(Korea)、MK68526 (China)、MH021660.1(India)、MF480443.1(Korea)聚类到一支,相互遗传距离较近,且共同的结点位于中国地区旱獭埃希氏菌聚集分支结点前,推测中国地区旱獭埃希氏菌是由该分支进化而来。同时发现,出现的菌株全部为亚洲大陆的,没有发现其他大陆地区的踪迹,说明旱獭埃希氏菌更适宜生存的地区可能在亚洲大陆,且可能是旱獭埃希氏菌的发源地[17-20]。
由图 1还可以发现,在伤口埃希氏菌中,MF079358.1(China)、KX357823.1(India)、LT631770.1(Belgium)、LC040946.1(Pakistan)、AB681776.1(Japan)、JQ958880.1(France)、KR612005.1(India)、AF530476.1(Canada)在地域距离上相隔较远,彼此间遗传距离较大,但最终聚类为一支。从图中可以明显发现,分支产生的顺序在地理位置上有着一定的规律:东亚、南亚、西亚、欧洲、北美洲,说明伤口埃希氏菌的传播可能发源于亚洲,并且在传播过程中不断发生不同程度变异。
4 讨论
42株菌株分为四大分支[21],不同分支间集中分布明显,说明四个分支间存在较高保守性,且不同菌属可能朝着不同的方向进化[19]。大肠埃希氏菌与弗格森埃希氏菌分布为一支,相互之间同源性高,遗传距离近,与其他菌型亲缘关系较远,在共同进化中易发生变异,如 KF938590.1(Korea)、KJ806495.1(China)、KC986857.1(New Zealand)、MK729003.1(China)、NR 114042.1(Japan)、KJ726590.1(U.S.A),其进化程度与地理位置关系不大。在艾伯特埃希氏菌中,来自南极洲的两株在遗传关系上距离较近,同源性高达98.2%,来自中国的两株在遗传关系上距离却较远,表明艾伯特埃希氏菌的类型与生存环境条件有很大关联性,该菌型进化方向仍未突破地域,而基于16S rRNA序列分析,推测艾伯特埃希氏菌的起源可能是多源的,环境条件可能作为重要的进化压力,在艾伯特埃希氏菌长期演化过程中扮演重要角色[19];另外,来自中国的一株与南极的两株在遗传关系上距离较近,另一株中国的与来自澳大利亚的一株在遗传关系上距离较近,说明该菌型在亚洲与大洋洲、南极洲三大洲间可能存在着交叉进化。
在进化树上,4 株中国基因型旱獭埃希氏菌菌株与其他不同地区菌株分离明显,各自独立进化,且它们全部位于亚洲大陆,可以推测旱獭埃希氏菌更适宜生存的地区在亚洲大陆,研究中国此类菌株更有利于揭示旱獭埃希氏菌基因的遗传进化规律,伤口埃希氏菌在地域距离上相隔较远且株间分布较散,可能与伤口埃希氏菌极易感染伤口的特性有关[17-21]。
在同源性比较与分子进化树中,也有一些菌株散落在不同菌种的分支间,并且与周边菌株的同源性极高,除了基因变异,推测是由于一些特殊因素造成细菌传播,如人口流动、物种引进、候鸟迁徙、人工养殖和商贸交流。在进化树的分支中,接壤国家之间的菌株同源性相对较高,说明埃希氏菌在陆地之间传播的可能性更高,但是不接壤国家间也存在同源性极高的菌株,不排除埃希氏菌间接传播的可能。本研究在国内首先开展埃希氏菌属遗传演化关系分析,从分子水平阐明各个菌株流行分布规律,为更好预防埃希氏菌病传播提供理论依据。
