基于HPLC-Q-TOF-MS的非那雄胺原料杂质结构及来源分析*
2023-01-12曾庆花朱健萍卢日刚
邓 鸣,曾庆花,朱健萍,卢日刚
(广西壮族自治区食品药品检验所,广西南宁 530021)
非那雄胺(Finasteride)化学名称为N-叔丁基-3-氧代-4-氮杂-5α-雄甾-1-烯-17β-甲酰胺,分子式为C23H36N2O2。非那雄胺最早由美国默沙东公司开发研制,1991年获准上市。非那雄胺是一种合成的甾体激素化合物,它是雄激素睾酮代谢为双氢睾酮(Dihydrotestosterone,DHT)过程中的细胞内酶Ⅱ型5α还原酶的特异性抑制剂,具有抑制睾酮代谢的作用。良性前列腺增生(Benign Prostate Hyperplasia,BPH,也称为前列腺肥大)有赖于体内有效的双氢睾酮的含量,即取决于前列腺中睾酮向DHT的转化。非那雄胺能有效降低血液和前列腺内的DHT浓度。目前非那雄胺制剂有5 mg和1 mg两种规格,前者适应证为治疗和控制良性前列腺增生及相关症状,后者用于治疗男性脱发[1,2]。
经查阅各国药典,非那雄胺被《中华人民共和国药典:2020年版·二部》[3]、EuropeanPharmacopoeia(《欧洲药典》)10.0版[4]、UnitedStatesPharmacopeia(《美国药典》)2022年版[5]收载,均采用高效液相色谱(High Performance Liquid Chomatography,HPLC)法检查有关物质。其中《欧洲药典》10.0版该品种项下给出了杂质A、杂质B、杂质C的结构。《中华人民共和国药典:2020年版·二部》中杂质I即为《欧洲药典》10.0版中的杂质A,在以下的叙述中该杂质均以杂质A表示。
文献[6-9]采用高效液相色谱法分离非那雄胺杂质。近年来,液质联用技术已日臻成熟,并被广泛应用于药物中杂质的结构鉴定及来源分析[10-14]。本研究拟采用高效液相色谱-四极杆飞行时间质谱法(High Performance Liquid Chromatography- Quadrupole Time of Flight Mass Spectrometry,HPLC-Q-TOF-MS)研究非那雄胺的杂质,并通过分析非那雄胺的裂解特征,与已知杂质对照品比对,通过强制降解试验等途径对其中的主要杂质进行结构推断,推测杂质可能的来源,以期为药品杂质控制和工艺优化提供参考依据。
1 材料与方法
1.1 仪器与试药
Waters Xevo G2 Q-TOF液质联用仪(美国Waters公司),MILLI-Q去离子水发生器(美国Millipore公司),XS205DU 电子天平[梅特勒托利多科技(中国)有限公司]。
非那雄胺对照品(中国食品药品检定研究院,批号100611-201503,含量99.7%),杂质I (杂质A)(中国食品药品检定研究院,批号420023-201501),杂质B(Toronto Research Chemicals Inc.,批号Y11M10W86170),杂质C (Toronto Research Chemicals Inc.,批号Y11M10W86171)。
非那雄胺原料来源于国内两个厂家,A厂家及B厂家各1批。乙腈为色谱纯试剂,水为超纯水,其他试剂为分析纯。
1.2 溶液的制备
混合对照品溶液:称取非那雄胺对照品、杂质A、杂质B、杂质C对照品各约1 mg,置50 mL量瓶中,加50%乙腈溶液溶解并稀释至刻度,摇匀。
供试品溶液:称取非那雄胺原料(A厂家或B厂家)约10 mg,置10 mL量瓶中,加50%乙腈溶液溶解并稀释至刻度,摇匀。
1.3 强制降解试验
取非那雄胺原料(A厂家)约50 mg,精密称定,置于10 mL量瓶中,加50%乙腈溶液溶解并稀释至刻度,摇匀,作为贮备液。
精密量取贮备液1 mL,置于5 mL量瓶中,加1 mol/L盐酸溶液1 mL,放置5 h,加入1 mol/L 氢氧化钠溶液1 mL中和,加50%乙腈溶液稀释至刻度,摇匀,滤过,即得酸破坏溶液。
精密量取贮备液1 mL,置5 mL量瓶中,加1 mol/L氢氧化钠溶液1 mL,放置2 h,加入1 mol/L 盐酸溶液1 mL中和,加50%乙腈溶液稀释至刻度,摇匀,滤过,即得碱破坏溶液。
精密量取贮备液1 mL,置5 mL量瓶中,加30%过氧化氢溶液1 mL,放置1 h,加入50%乙腈溶液稀释至刻度,摇匀,滤过,即得氧化破坏溶液。
取非那雄胺原料(A厂家)适量,置120℃烘箱中加热5 h,放冷,称取约10 mg置10 mL量瓶中,加入50%乙腈溶解并稀释至刻度,摇匀,滤过,即得高温破坏溶液。
取非那雄胺原料(A厂家)适量,置4 500 lx光照条件下放置48 h,称取约10 mg置10 mL量瓶中,加入50%乙腈溶解并稀释至刻度,摇匀,滤过,即得强光破坏溶液。
1.4 色谱与质谱条件
1.4.1 色谱条件
色谱柱:资生堂Capell-MGⅡ C18(5 μm,4.6 mm×250 mm);流动相为乙腈∶水(45∶55),流速:1.0 mL·min-1;柱后分流,检测波长:210 nm;柱温:30℃;进样量:2 μL。
1.4.2 质谱条件
离子化方式:ESI(+/-);数据采集质荷比(m/z) 80-800;干燥气温度:350℃;脱溶剂气(N2)流速:6.0 L·min-1;碰撞电压:10-30 eV;毛细管电压:4.5 kV。
2 结果与分析
2.1 非那雄胺高效液相色谱图及杂质来源分析
将非那雄胺供试品溶液、强制降解溶液进样后,主峰与各杂质峰分离良好。供试品溶液中主要有15个峰,按保留时间顺序命名为杂质1-15 (图1)。强制降解试验结果表明,非那雄胺在氧化、光照、高温条件下较稳定,未产生明显的降解产物;强酸破坏时杂质4含量明显增大,杂质6和杂质8含量有小幅度增大;强碱破坏时杂质2增大;高温破坏时杂质C含量有小幅度增大。因此杂质1,3,5,7,9,10,11,12,14,15为工艺杂质,杂质2,4,6,8和C可能是降解杂质,也可能是工艺杂质。通过分析杂质的来源,提示企业应优化生产工艺,并在贮藏及运输过程中控制温度,减少非那雄胺降解反应的发生。
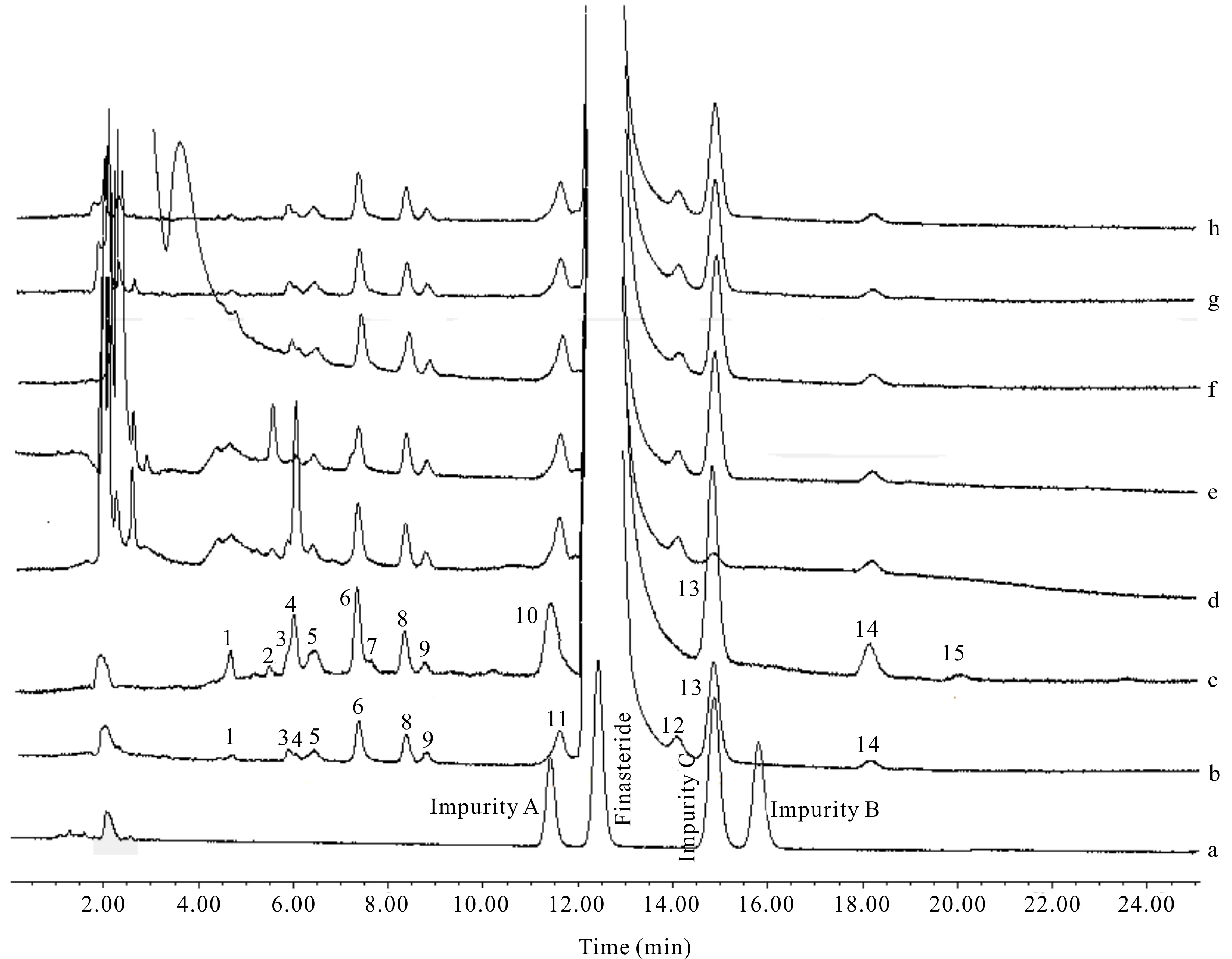
a:Mixed solution of reference substances;b:Finasteride (A manufacturer);c:Finasteride (B manufacturer);d:Acidic hydrolysis;e:Alkaline hydrolysis;f:Oxidative degradation;g:Photolytic degradation;h:Thermo degradation;number 1-15:Impurity 1-15图1 对照品混合溶液、原料供试液及强制降解溶液高效液相色谱图Fig.1 HPLC chromatograms of mixed reference solution,raw test solution and forced degradation solution
2.2 非那雄胺主要杂质的结构鉴定
杂质结构与主药往往具有相关性,通过分析非那雄胺二级质谱裂解规律并结合破坏试验推测杂质1,2,5,6,8,10,13,14等8种杂质的结构,其中杂质10,13分别对应《欧洲药典》10.0版的特定杂质A、C,其他7种杂质因含量过低或受其他因素干扰,未推断出结构。为了确证所推断的机理,对推断的杂质均进行了高分辨数据分析(表1) 。
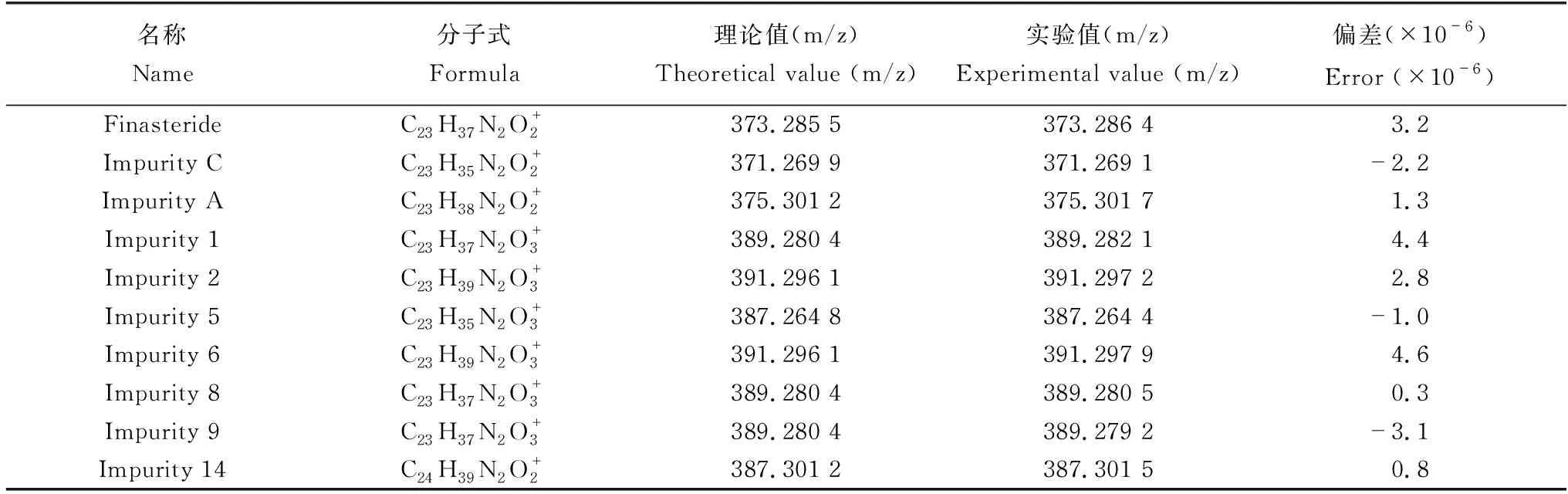
表1 杂质准分子离子理论值与实验值的偏差计算Table 1 Deviation calculation of theoretical values and experimental values of the impurities in MS2
2.2.1 非那雄胺
非那雄胺准分子离子m/z为373.286 4,其二级质谱碎片中主要的碎片峰为侧链断裂后,脱掉分子量为56的叔丁基形成m/z为317.223 4的碎片离子,其侧链还可继续断裂生成m/z分别为299.214 9,282.186 6,272.204 3的碎片离子,母核结构中的氮杂环开环后脱去质量数为68的C4H4O后可形成m/z为305.259 9的碎片离子,该碎片离子进一步脱去侧链的基团后可生成m/z分别为187.148 6,215.142 0的碎片离子,但丰度均较少,其二级扫描质谱图及裂解途径见图2。
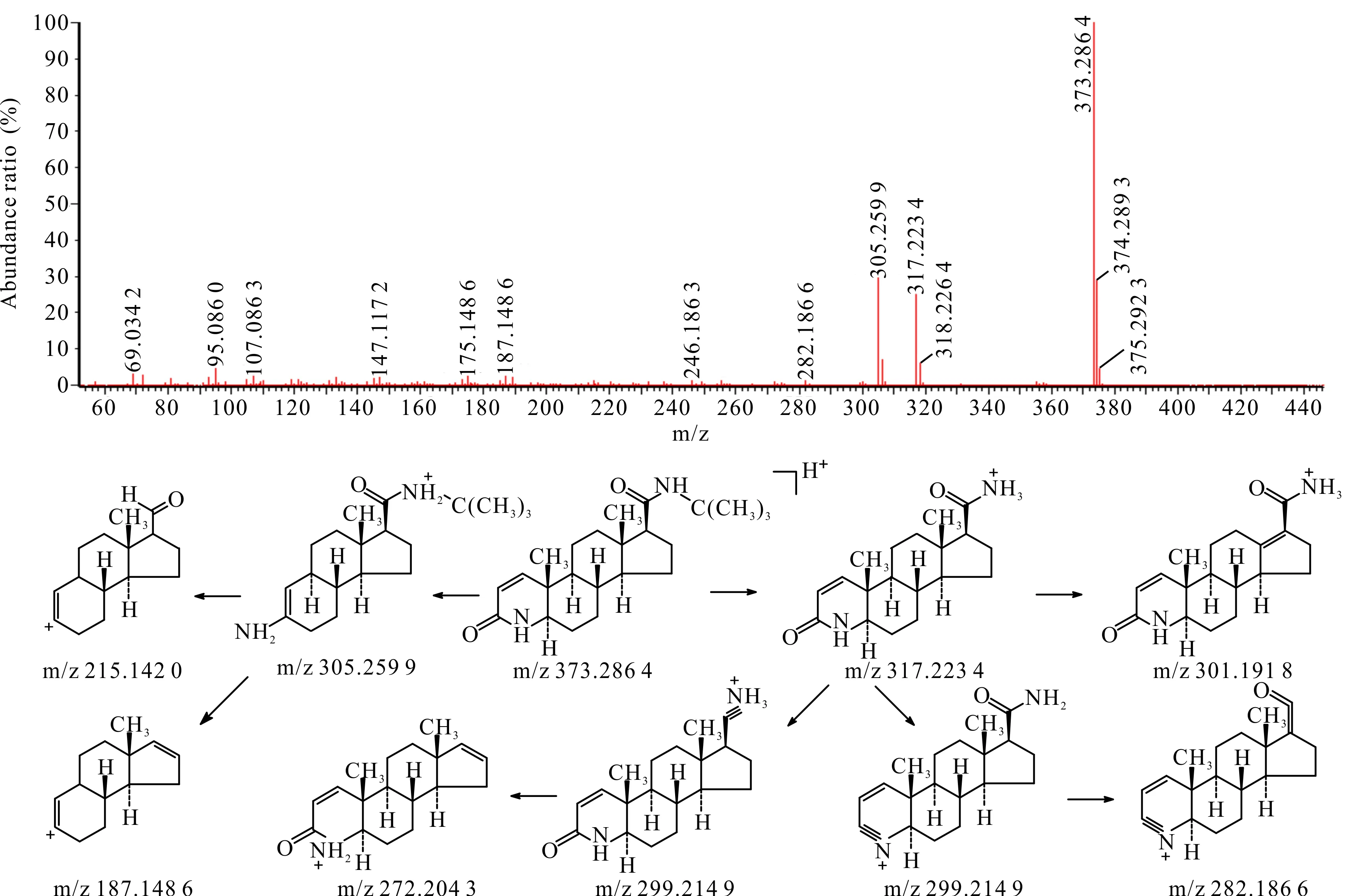
图2 非那雄胺[M+H]+二级扫描质谱图及裂解途径Fig.2 [M+H]+ secondary scanning mass spectrum and fragmentation pathway of finasteride
2.2.2 杂质13
在HPLC-UV-Q-TOF-MS中,杂质13的液相保留时间、紫外光谱图、准分子离子(m/z为371.269 1)及二级质谱均与杂质C对照品一致,因此确证杂质13为杂质C。
杂质C为非那雄胺合成过程的中间产物。杂质C的裂解方式与非那雄胺类似,侧链断裂后可形成m/z分别为315.206 3,298.184 5,270.185 3,280.169 6的碎片离子,母核结构中的氮杂环开环后脱去C4H4O后可形成m/z为303.247 3的碎片离子。母核结构中的C5与C6为双键,易脱去C上的甲基,形成具有共轭双键的碎片结构,该碎片离子的m/z为355.236 9。杂质C二级扫描质谱图及裂解途径见图3。
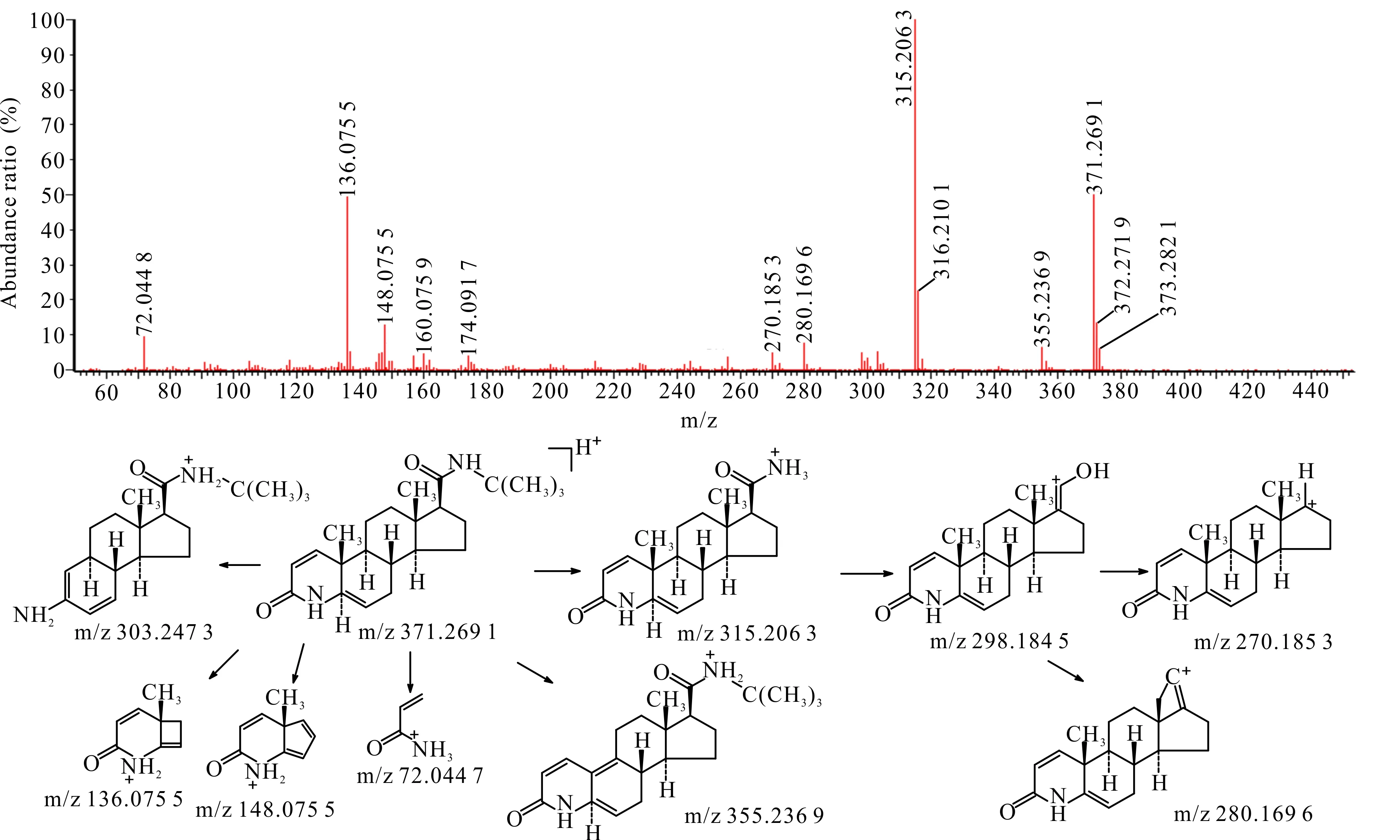
图3 杂质13 [M+H]+二级扫描质谱图及裂解途径Fig.3 Impurity 13 [M+H]+ secondary scanning mass spectrum and its fragmentation pathway
2.2.3 杂质10
杂质10的液相保留时间、紫外光谱图均与《欧洲药典》10.0版收载的杂质A对照品一致。在HPLC-UV-Q-TOF-MS中该杂质的准分子离子(m/z为375.301 7)及其二级质谱中各子离子的质荷比、丰度也与杂质A对照品基本一致,因此确认杂质10为杂质A。
杂质A的裂解方式与非那雄胺类似,侧链断裂后形成的碎片离子以及母核结构中的氮杂环开环后形成碎片离子。杂质A二级扫描质谱图及裂解途径见图4。
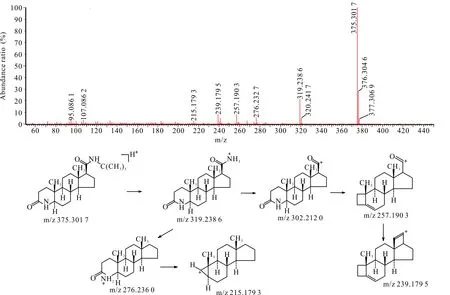
图4 杂质10 [M+H]+二级扫描质谱图及裂解途径Fig.4 Impurity 10 [M+H]+ secondary scanning mass spectrum and its fragmentation pathway
2.2.4 杂质1
杂质1在HPLC-UV-Q-TOF-MS中的准分子离子m/z为389.282 1,较非那雄胺的准分子离子质荷比约多16,推测为多一个氧原子,其二级谱图中出现了较母离子少72的碎片离子,推测非那雄胺结构中的叔丁基氧化为叔丁醇。杂质1的裂解方式与非那雄胺类似,二级图谱中有侧链断裂后形成的碎片离子以及母核结构中的氮杂环开环后形成碎片离子,此处不再赘述。杂质1二级扫描质谱图及裂解途径见图5。
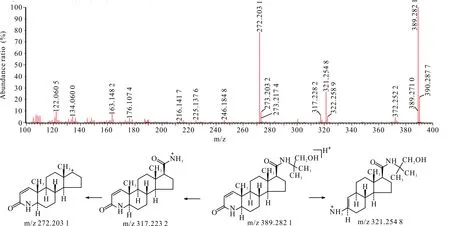
图5 杂质1 [M+H]+二级扫描质谱图及裂解途径Fig.5 Impurity 1 [M+H]+ secondary scanning mass spectrum and its fragmentation pathway
2.2.5 杂质2
杂质2在HPLC-UV-Q-TOF-MS中的准分子离子m/z为391.297 2,与非那雄胺的m/z相差约18,且其二级质谱出现M+H-56的碎片峰,推测该杂质为非那雄胺的氮杂环水解开环后生成的杂质。其二级质谱出现m/z为373.288 2的碎片为羧酸与氨基重新环合生成的。杂质2二级扫描质谱图及裂解途径见图6。

图6 杂质2 [M+H]+二级扫描质谱图及裂解途径Fig.6 Impurity 2 [M+H]+ secondary scanning mass spectrum and its fragmentation pathway
2.2.6 杂质3
杂质3在HPLC-UV-Q-TOF-MS中的准分子离子m/z为387.264 4,与非那雄胺的m/z相差约14,推测可能的分子式为C23H34N2O3,其二级质谱出现M+H-54、M+H-68的碎片峰,未出现M+H-56的碎片峰,因此推测杂质3具有与非那雄胺相同的氮杂环,而侧链部分结构发生改变。另外,杂质3二级质谱中的碎片离子与非那雄胺均不一致,推测其母环结构与非那雄胺不同,根据现有的信息无法推测碎片离子的组成,因此未能推断其结构。
2.2.7 杂质4
杂质4在HPLC-UV-Q-TOF-MS中的准分子离子m/z为389.278 8,与非那雄胺的m/z相差约16,推测可能的分子式为C23H36N2O3,其二级质谱出现M+H-56的碎片峰,未出现M+H-68的碎片峰,因此推测杂质4母核结构与非那雄胺不一致。根据现有的信息无法推测碎片离子的组成,因此未能推断其结构。
2.2.8 杂质5
杂质5在HPLC-UV-Q-TOF-MS中的准分子离子m/z为387.264 4,与非那雄胺的m/z相差约14,其二级质谱出现M+H-56、M+H-68碎片峰,未出现M+H-73的碎片峰,而出现了M+H-87,因此杂质5侧链上的叔丁基结构未变,而N上多一个甲基。杂质5二级扫描质谱图及裂解途径见图7。

图7 杂质5 [M+H]+二级扫描质谱图及裂解途径Fig.7 Impurity 5 [M+H]+ secondary scanning mass spectrum and its fragmentation pathway
2.2.9 杂质6
杂质6在HPLC-UV-Q-TOF-MS中的准分子离子m/z为391.297 9,与非那雄胺的m/z相差约18。杂质6二级质谱出现M+H-56、M+H-73碎片峰,未出现M+H-68的碎片峰,因此推测杂质6为非那雄胺母核氮杂环发生了变化。杂质6二级扫描质谱图及裂解途径见图8。

图8 杂质6 [M+H]+二级扫描质谱图及裂解途径Fig.8 Impurity 6 [M+H]+ secondary scanning mass spectrum and its fragmentation pathway
2.2.10 杂质8
杂质8在HPLC-UV-Q-TOF-MS中的准分子离子m/z为389.280 5,与非那雄胺的m/z相差约16。杂质8二级质谱出现M+H-56、M+H-68碎片峰,而其他碎片离子均与非那雄胺不一致,由于N原子可能会被氧化,因此推测杂质8为非那雄胺母核氮杂环形成了氮氧化物。杂质8二级扫描质谱图及裂解途径见图9。
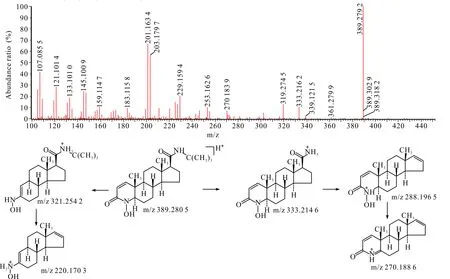
图9 杂质8 [M+H]+二级扫描质谱图及裂解途径Fig.9 Impurity 8 [M+H]+ secondary scanning mass spectrum and its fragmentation pathway
2.2.11 杂质9
杂质9在HPLC-UV-Q-TOF-MS中的准分子离子m/z为389.279 2,与非那雄胺的m/z相差约16。杂质6二级质谱出现M+H-56的碎片峰,未出现M+H-68的碎片峰,因此推测杂质9为非那雄胺母核氮杂环发生了变化,可能为双键氧化成酮基。杂质9二级扫描质谱图及裂解途径见图10。
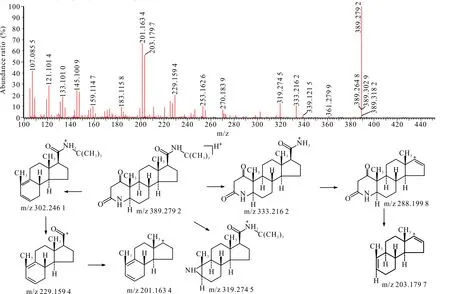
图10 杂质9 [M+H]+二级扫描质谱图及裂解途径Fig.10 Impurity 9 [M+H]+ secondary scanning mass spectrum and its fragmentation pathway
2.2.12 杂质11
杂质11在HPLC-UV-Q-TOF-MS中的准分子离子m/z为371.270 0,与非那雄胺杂质C的分子量一致。杂质11的二级质谱与杂质C部分一致,都出现m/z分别为298,280,270的碎片峰,因此推测杂质11为非那雄胺母核碳碳双键位置不同,已有信息无法判断双键的位置,因此未能推断其结构。
2.2.13 杂质14
杂质14在HPLC-UV-Q-TOF-MS中的准分子离子m/z为387.301 5,与非那雄胺的m/z相差约14。杂质6二级质谱出现M+H-68、M+H-70的碎片峰,未出现M+H-56的碎片峰,因此推测杂质14为非那雄胺侧链N原子被2-甲基丁基取代。杂质14二级扫描质谱图及裂解途径见图11。
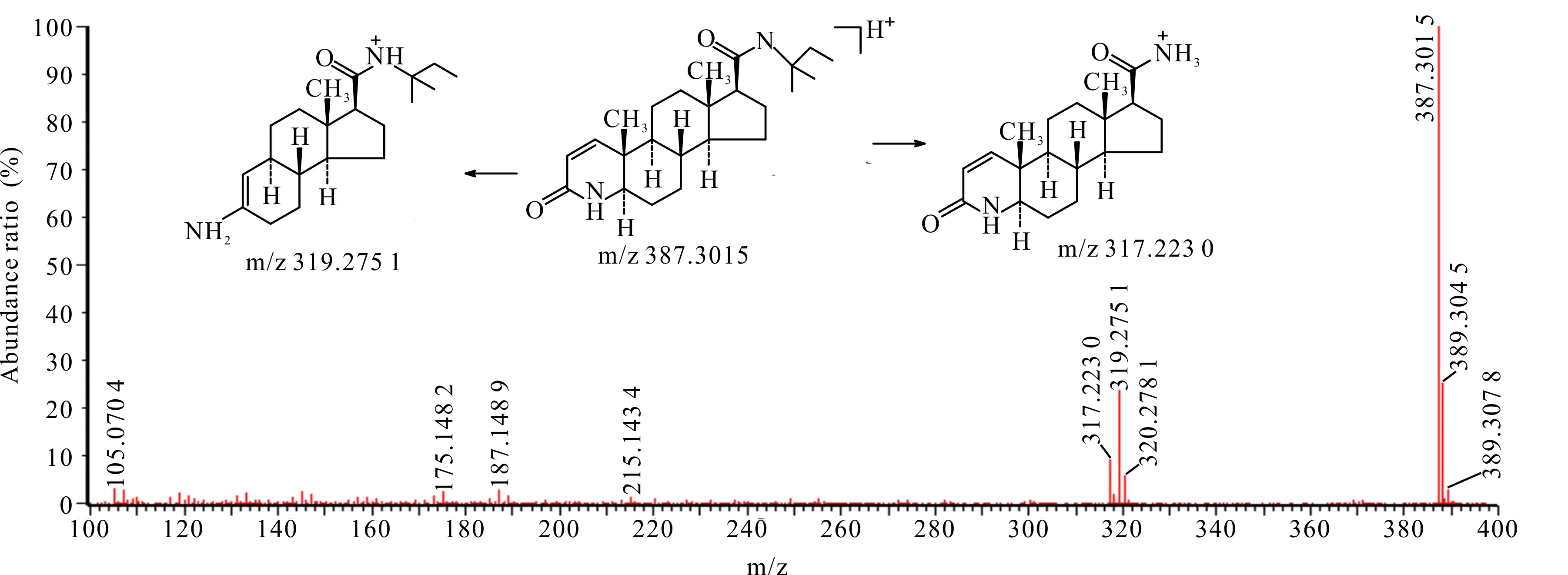
图11 杂质14 [M+H]+二级扫描质谱图及裂解途径Fig.11 Impurity 14 [M+H]+ secondary scanning mass spectrum and its fragmentation pathway
2.2.14 其他杂质
杂质7,12和15由于峰响应较低,未能推断其结构。
3 结论
本文采用HPLC-TOF-MS对两家非那雄胺原料生产企业的产品进行比较分析,发现两家生产的非那雄胺其杂质的种类及含量存在一定差异;鉴定出8种杂质的结构,并推测了部分杂质的来源,可为非那雄胺杂质控制和工艺优化提供参考依据。
