藏柳茶多糖的提取、结构解析及抗氧化活性分析
2023-01-12杨许花张竞文刘红海高丹丹丁功涛宋礼马姝雯
杨许花,张竞文,刘红海,高丹丹*,丁功涛,宋礼,马姝雯
(1.西北民族大学生命科学与工程学院,甘肃兰州 730124)(2.西北民族大学生物医学研究中心,甘肃兰州730030)(3.甘肃烟草工业有限责任公司,甘肃兰州 730050)(4.甘南牦牛乳研究院,甘肃甘南 747000)
藏柳茶(Sibiraea laexigata(L.)Maxim),又名窄叶鲜卑花,隶属蔷薇科鲜卑花属,常以其嫩叶、枝用作茶饮而成为藏族民间传统珍贵药材,主要分布于我国青海南部、四川西部、甘肃东南部和云南北部及西藏等高海拔地区[1]。现代药理学研究表明,藏柳茶主要有效成分包括三萜类化合物、黄酮以及多糖类,因柳茶特有的生理活性和天然来源的安全性使其在保健品和功能性食品中具有强大的应用潜力。多糖又称多聚糖,是一种天然药物成分,由许多相同或不同的单糖以α-或β-糖苷键构成的化合物,是生物体维持生命活动的主要能源物质[2]。柳茶多糖(Sibiraea angustataPolysaccharides,SAPs)作为藏柳茶主要的生物活性物质,具有清热助消化、调节脂代谢、抗氧化、抗肿瘤及增强机体免疫活性[3]等药用价值。目前关于藏药柳茶的研究主要以水提物为主,鲜有关于SAPs 提取工艺优化及结构表征和抗氧化活性的研究报道,本文首次采用聚乙二醇(Polyethylene Glycol,PEG)-超声酶法辅助提取(Ultrasonic-Assisted Enzymatic Extraction,UAEE)技术优化萃取SAPs,该法较常用传统水提醇沉[4]不仅引入超声波辅助缩短提取时间,PEG 试剂提供-OH 基团以加强多糖的相互作用和酶解双重作用下提高多糖在PEG 溶液中的溶解度以大大提高多糖提取率。
本研究以采自甘肃太子山国家级自然保护区柳茶为研究材料,拟在单因素试验的基础上,采用响应面试验筛选出PEG-UAEE 辅助提取SAPs 的最佳工艺参数,并对单糖组成、结构和抗氧化活性进行分析,以期为柳茶多糖工业化提取和产品的综合开发利用提供理论技术支持依据。
1 材料与方法
1.1 材料与试剂
藏柳茶:采于甘肃省太子山国家级自然保护区(102 °43 'E,36 °36 'N);果胶酶及纤维素酶,上海源叶生物科技有限公司;聚乙二醇,天津市大茂化学试剂厂;1,1-二苯基-2-三硝基苯肼(DPPH),Sigma 公司;三氟乙酸及单糖标品,美国Sigma 公司。其余均为国产分析纯试剂。
1.2 主要仪器设备
安捷伦1260 高效液相,美国Agilent 公司;650 红外光谱仪,天津港东科技股份有限公司;ZEISS EVO18扫描电镜,德国卡尔蔡司;Heraeus Multifuge X1R 高速冷冻离心机,赛默飞世尔科技有限公司;Fisher 酶标仪,美国Thermo 公司;LGJ-100F 真空冷冻干燥机,北京松源华兴科技发展有限公司;ZWYR-2401 恒温培养振荡器,上海智城分析仪器制造有限公司;SB-500DTY超声清洗器,宁波新芝生物科技有限公司。
1.3 试验方法
1.3.1 主要仪器设备
(1)提取工艺流程
新鲜原料→干燥→粉碎→过60 目筛→石油醚脱脂、φ=85%乙醇脱色→称取3 g 柳茶叶干粉→聚乙二醇、超声酶法辅助提取→离心(5 000 r/min,10 min)取上清液→蒸发浓缩至1/3→4 倍体积乙醇醇沉→4 ℃静置12 h→离心(8 000 r/min,5 min)→冷冻干燥→柳茶粗多糖
(2)多糖除蛋白、脱色操作流程
柳茶粗多糖液(0.1 g/mL)→3 倍量Sevage 试剂(正丁醇:氯仿=1:5,V/V)除蛋白→0.06%(m/m)活性炭脱色(40 ℃水浴1 h)→振荡混匀30 min→透析36 h(分子量3 500 u)→蒸发浓缩至1/3→4 倍量乙醇醇沉→离心(8 000 r/min,5 min)→淋洗(无水乙醚、丙酮、无水乙醇多次)→冷冻干燥→精制SAPs→苯酚-硫酸法测多糖含量。
1.3.2 SAPs 提取率及含量计算
多糖提取率计算如公式(1)所示:

式中:
A——多糖得率,%;
m——冻干柳茶多糖质量,g;
M——柳茶干粉质量,g。
采用苯酚-硫酸法[5]测定其多糖含量。以D-葡萄糖为对照制作标准曲线,在质量浓度0.1~0.6 mg/mL 范围内绘制标准曲线,其回归方程y=0.629 1x-0.005 9,R2=0.999 2,计算柳茶多糖含量。
1.3.3 PEG 分子量和浓度对SAPs 提取率的影响
考察不同PEG 分子量(200、400、600、800、1 000 u)和不同PEG 质量分数(15%、20%、25%、30%、35%)对多糖提取率的影响。
1.3.4 单因素条件优化
以3.0 g 柳茶叶干粉,液料比15 mL/g,纤维素酶:果胶酶=1:2 酶配比为基础实验条件。酶添加量(1.0%、1.5%、2.0%、2.5%、3.0%,m/m);pH 值(3.0、4.0、5.0、6.0、7.0);超声功率(200、250、300、350、400 W);超声时间(1.0、1.5、2.0、2.5、3.0 h);超声温度(40、50、60、70、80 ℃)对SAPs 提取率的影响,平行测定3 次。
1.3.5 响应面优化
在单因素试验基础上,以粗多糖得率(Y)为响应值,酶添加量(A)、pH(B)、超声时间(C)、超声温度(D)为自变量,进行四因素三水平Box-Behnken 响应面试验设计(BBD),因素及水平设计见表1。响应面水平见下表3。
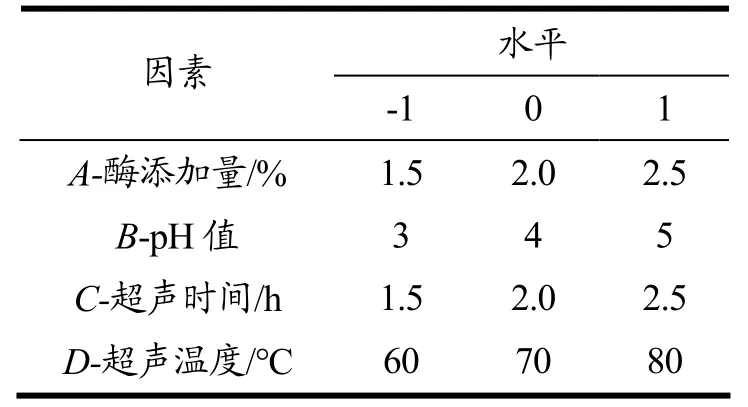
表1 响应面试验设计因素水平表Table 1 Factors of response surface test design
1.3.6 精制SAPs 的纯化
参考宫春宇等[6]方法,配制精制0.1 g/mL SAPs 溶液过0.45 μm 微孔滤膜后,依次过100、30、10 ku 超滤膜进行纯化,以获得相对分子质量<10 ku 的SAPs溶液,并醇沉冻干备用。
1.3.7 分子量分布测定
参考Wang 等[7]方法,配制1.0 mg/mL 纯化SAPs,采用高效凝胶渗透色谱法(High Performance Gel Permeation Chromatography,HPGPC)测定SAPs 分子量。选用UltrahydrogelTMLinear Column(300×7.8 mm,8 μm)色谱柱;调节柱温40 ℃、流速0.8 mL/min 及进样体积为20 μL;流动相为0.1 mol/L NaNO2溶液。使用Dextran(Mw:1、5、10、21、40、84 ku)作为校准标准,得到标准曲线方程为logMw=-0.649 3x+6.156 4(R2=0.998 7),以计算纯化后SAPs 样品的相对分子量。
1.3.8 单糖组成分析
1.3.8.1 薄层层析[8]
展开剂:正丁醇:异丙醇:乙酸乙酯:醋酸:吡啶:水=35:60:100:35:30:30(V/V)。
染色剂:0.9 mL 苯胺和1.66 g 邻苯二甲酸混合溶解,加正丁醇至100 mL。
取木糖、葡萄糖、阿拉伯糖、果糖、甘露糖、半乳糖、鼠李糖等7 种标品和多糖水解产物点于薄层析纸以展开,取出吹干后均匀喷洒显色剂于100 ℃烘至斑点显色清晰。
1.3.8.2 HPLC 色谱法分析单糖组成
参考Liang 等[9]试验方法,采用PMP 柱前衍生高效液相色谱(HPLC-PMP)分析SAPs 的单糖组成。
(1)多糖水解及衍生化:精确称取10.00 mg 粗多糖样品于螺口管中,加入2 mol/L 三氟乙酸(TFA)溶液5 mL,氮封后于110 ℃水浴水解5 h;冷却后用3 mol/L NaOH 调节pH 值为7.0,超纯水定容至10 mL 后离心得多糖水解产物。各取0.2 mL 的0.5 mol/L 1-苯基-3-甲基-5-吡唑啉酮(PMP)/甲醇溶液和0.3 mol/L NaOH加入到0.2 mL多糖水解液中,漩涡混匀后70 ℃水浴1 h,冷却后加0.3 mol/L HCl溶液0.2 mL和1 mL氯仿混匀萃取,离心取上清液过0.22 μm 滤膜做单糖组成分析。
(2)色谱条件:采用Agilent ZORBAX Eclipse XDB-C18 色谱柱(4.6×250 mm,5 μm);流动相:A为0.02 mol/L 的磷酸盐缓冲液(pH 值6.8),B 为乙腈;柱温28 ℃,流速:0.8 mL/min;进样量:5 μL,检测波长:250 nm;洗脱梯度如表2 所示。
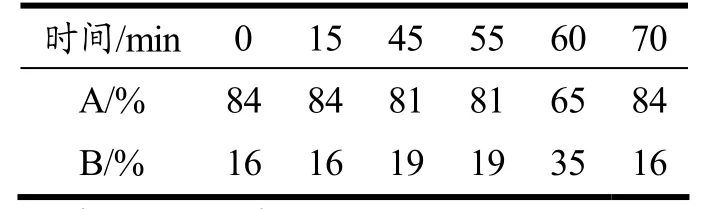
表2 流动相梯度Table 2 Mobile phase gradient
1.3.9 红外光谱分析
取适量纯化冻干SAPs 粉末,使用KBr 压片法研磨均匀压片后对样品在4 000~400 cm-1范围内进行傅立叶红外光谱分析(FT-IR)并记录[10]。
1.3.10 核磁共振氢谱分析
参考Wang 等[11]方法,取20 mg 纯化冻干SAPs 完全溶于0.5 mL D2O以制备(Nuclear Magnetic Resonance,NMR)样品,经Bruker AVANCE III HD 400 光谱仪在80 ℃下300 MHz 运行1 h 得到1H NMR 光谱。
1.3.11 扫描电镜分析
参考Nuerxiatia 等[12]试验方法,用导电胶将纯化冻干SAPs 固定在样品台上,真空喷金处理后在20.00 kV下用德国蔡司钨灯丝扫描电镜(ZEISS EVO18)进行扫描,观察不同倍数下的外貌形态。
1.3.12 体外抗氧化活性分析
分别配制浓度为0.0、0.2、0.4、0.6、0.8、1.0 mg/mL的SAPs 待测溶液,以Vc 作为阳性对照,在517 nm 波长下测定SAPs 对DPPH·清除能力[13];在510 nm 波长下测定SAPs 对·OH 清除能力[14];在320 nm 波长下测定SAPs 对·清除能力[15];每组实验重复测定3 次,取平均值。
1.4 统计分析
所有统计分析均采用SPSS 软件进行差异显著性分析,数据结果均以3次实验数据的平均值±标准误差表示。此外,通过单因素方差分析分析组间差异,然后进行Duncan检验。p<0.05和p<0.01被认为具有统计学意义。
2 结果与分析
2.1 PEG 分子量和质量分数对SAPs 提取率的影响
PEG 试剂可提供较充足的-OH 基团以加强多糖的相互作用而形成氢键,从而提高多糖在PEG 溶液中的溶解度[16]。试验以30%(m/m)PEG 溶液(PEG-200、PEG-400、PEG-600、PEG-800、PEG-1000)分别作SAPs 提取溶剂。由图1a 可知,提取溶剂选择PEG-400 水溶液时SAPs 达最佳得率9.85%,后随着PEG 分子量的持续提高,多糖得率出现下降趋势。PEG 分子量不同,对萃取溶剂的扩散系数、粘度和极性产生的影响存在差异,本试验中PEG-400 较其他分子量更具有较大极性进行传质。
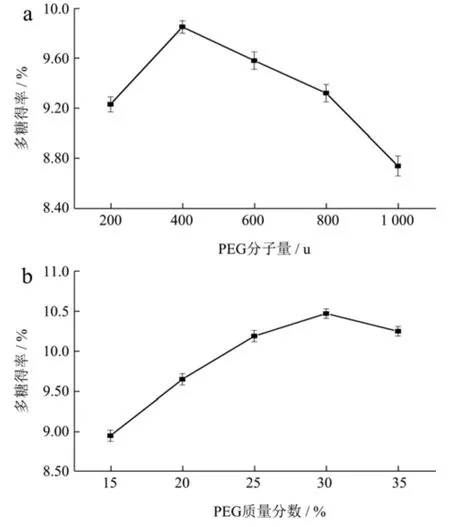
图1 PEG 分子量(a)、PEG 质量分数(b)对柳茶多糖得率的影响Fig.1 PEG molecular weight (a),PEG concentration (b) on the yield of SAPs
不同质量分数的PEG 萃取溶剂决定着溶液体系的粘度大小,进而影响多糖得率的高低[17]。由图1b 可知,PEG 质量分数在15%~30%时,柳茶多糖得率呈现明显的浓度依赖性增加,当质量分数达到30%时,多糖得率达到最大值10.47%,当质量分数大于30%时,多糖得率呈下降趋势。Zhou 等[18]采用质量分数30%PEG-400 水溶性溶剂提取石榴皮多糖,得到多糖产量最高达7.65%,可能是由于溶剂浓度的增加改变了体系的耗散因子,利于多糖的提取效率;当PEG 浓度多高时导致粘度过高而多糖得率下降。因此,PEG-400 提取SAPs 的最佳浓度为30%。
2.2 超声酶法提取
酶浓度的提高可以有效的增加酶与底物的结合面积,以使多糖提取率得到提高[19]。由图2a 可知,酶质量分数在1%~2%时,多糖提取率以剂量效应不断增加,当酶质量分数增加至2%时,多糖提取率提高1.06%;当酶质量分数超过2%后,由于酶的饱和度和底物有限,多糖提取率趋于平稳,这与Zhang 等[20]使用相同复合酶提取银杏叶多糖中提取率的趋势相似。在科学、经济性生产原则下选择酶质量分数为2%。
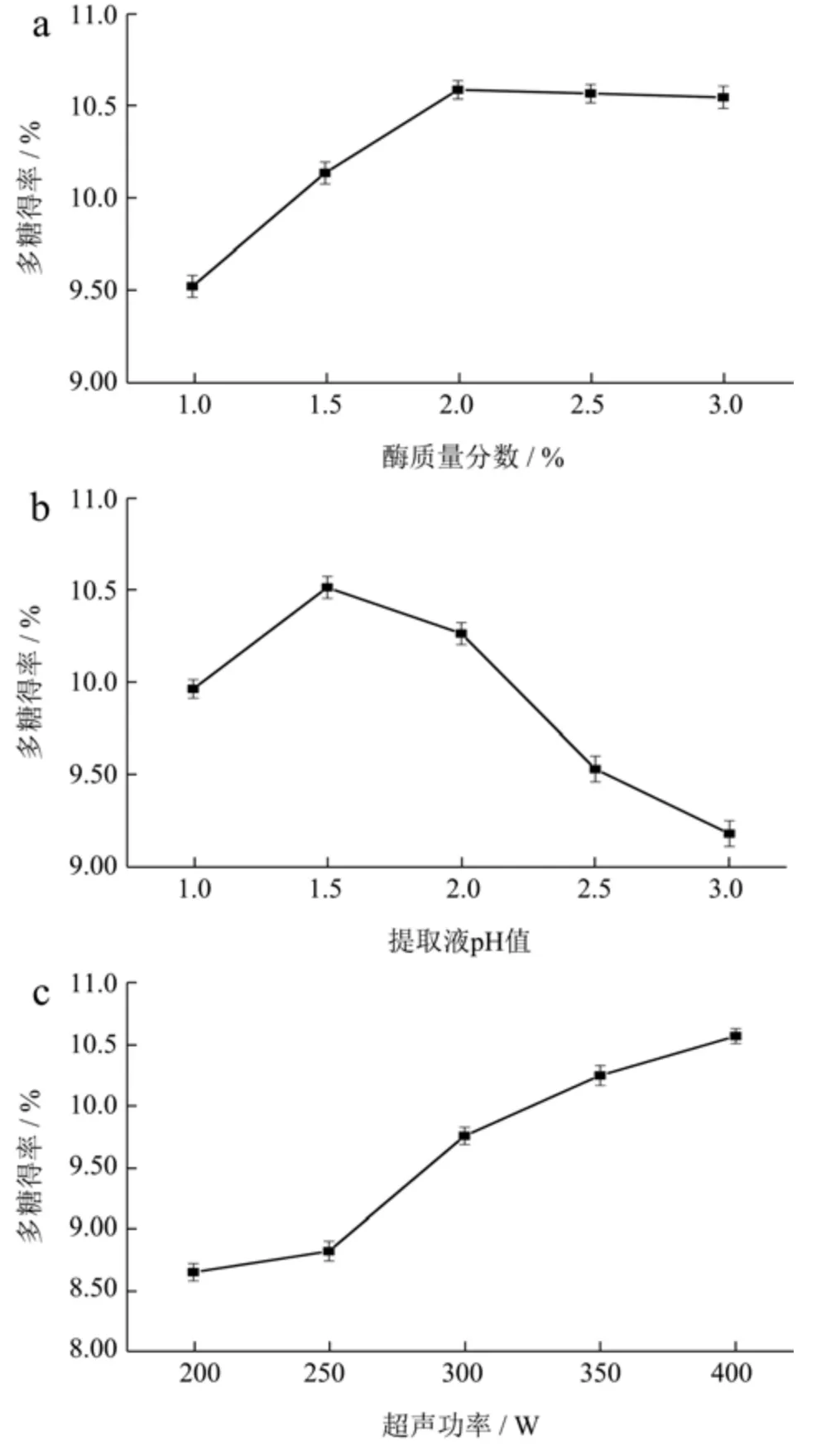
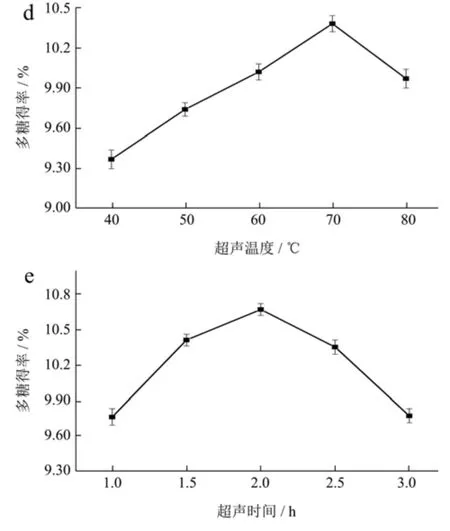
图2 酶添加量(a)、提取液pH 值(b)、超声功率(c)、超声温度(d)和超声时间(e)对柳茶多糖得率的影响Fig.2 Amount of adding enzyme (a),extraction pH (b),Ultrasonic power (c),Ultrasonic temperature (d) and Ultrasound time (e) on the yield of SAPs
提取液pH 值主要通过影响酶活性或调节多糖酸碱性来提高多糖得率[21]。由图2b 可知,提取液pH 值在3~7 范围内,多糖的提取率随pH 值的不断提高呈升后降的现象,当提取液pH 值等于4 时,多糖的提取率达最佳10.52%,Zhai 等[22]研究果胶酶用量为0.7%时不同提取液pH 值对石榴皮多糖产量的影响发现,多糖产量先随提取液pH 值的增加而增加,当pH 值超过5 时,可能由于SAPs 的主要成分是酸性多糖且试验中果胶酶最佳酶活pH 值为3.5,pH 值持续升高使的酸性多糖不能继续溶出且酶活降低。因此,选择提取液pH 值为4 条件下多糖提取率最高。
超声功率的不断提高产生的空化作用会引起高温高压环境使细胞破碎,多糖被有效释放溶解于萃取剂中,从而导致多糖得率提高[23]。如图1c 可知,超声功率为200~400 W 范围内,多糖提取率随超声功率的增大而不断提高,当超声功率为400 W 时,多糖得率达到最大10.56%。但随着超声功率不断提高会造成体系压力过大而破坏多糖结构,使多糖得率趋于平缓。考虑到仪器自身功率限制和实际生产的经济性,试验超声功率选择为400 W。
超声温度的升高可使柳茶粉软化以增加果胶酶和纤维素酶的酶解活性,加强酶对柳茶细胞结构的破坏,使多糖分子运动加快、接触并溶解于PEG 萃取剂[24]。由图1d 可知,多糖提取率在40~70 ℃范围内随着超声温度升高而迅速提高,在70 ℃时达到最高值10.38%,超声温度升高引起整个反应体系反应速度加快,加快多糖溶出。温度超过70 ℃后会加快萃取剂的损耗和多糖结构的破坏,使得多糖发生水解而得率降低[25]。综上,选择70 ℃为最佳超声温度提取。
超声时间的增加为超声波能提供充足有效作用时间破坏柳茶粉细胞壁,提高PEG 萃取剂萃取多糖[26]。由图1e 可知,超声时间在1.0~2.0 h 范围内,SAPs 得率呈现明显的浓度依赖性增加,当超声时间达到2.0 h时,多糖得率达到最大值10.67%,当时间超过2.0 h 后,多糖得率出现明显下降情况。可能因为高温高压状态持续时间过久或萃取液黏度增大,使超声波引起部分多糖发生水解或受黏度影响溶出率降低。选择超声时间2.0 h 为最佳超声时间提取。
2.3 响应面优化试验结果
2.3.1 回归模型的建立和方差分析
在单因素实验基础上,选择影响较显著的单因素:A(酶添加量)、B(pH 值)、C(超声时间)和D(超声温度)为相应因子,Y(粗多糖得率)为响应值,采用Design-Expert 8.0.6 软件对表1、表3 中的试验结果进行响应面分析,得到回归方程:
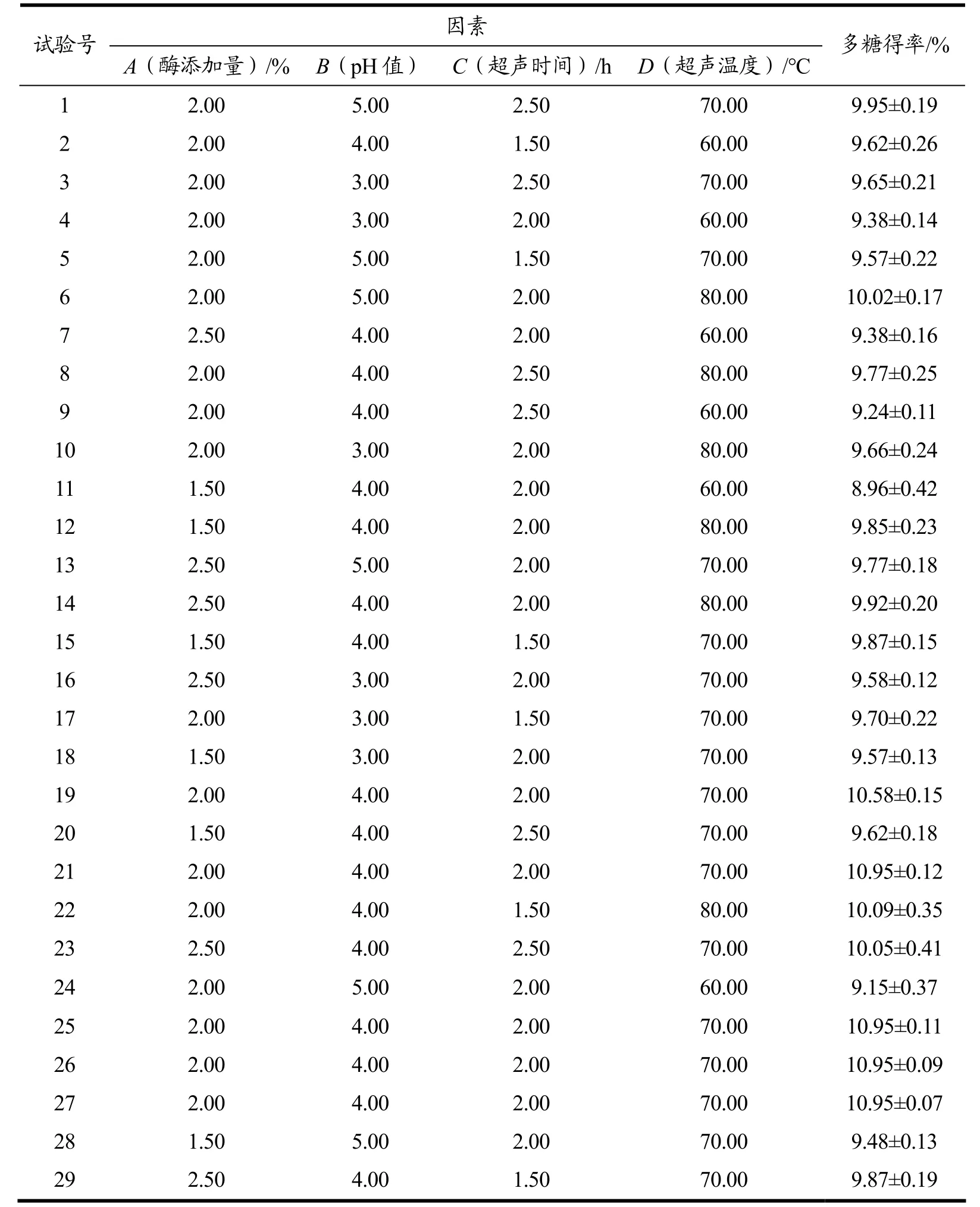
表3 柳茶多糖工艺优化响应面试验结果与分析Table 3 Response surface test results and analysis of SAPs
Y=10.88+0.10A+0.033B-0.037C+0.30D+0.070AB+0.11AC-0.087AD+0.11BC+0.15BD+0.015CD-0.60A2-0.66B2-0.47C2-0.71D2
实验方差结果分析如表4 所示,显著性检验表明该回归模型的拟合极显著(p<0.000 1),失拟项(p>0.05)不显著;模型与实际拟合较好(R2=0.983 1,R2adj=0.973 0),综上结果均可说明模型拟合度可靠性强,观察值和预测值之间的相关性良好,该模型可以用于预测和分析多糖最佳提取工艺[27]。该方差分析表明,线性系数D和二次系数(A2、B2、C2、D2)对柳茶多糖提取率影响极显著(p<0.000 1),线性系数A具有统计学意义(p<0.05)。

表4 回归方程方差分析及结果Table 4 Analysis of variance and results of regression equation

续表4
2.3.2 响应面分析及优化
影响SAPs 提取率的各因素交互作用3D 响应面和等高线如图3 所示。图3a~3f 分别表示酶添加量与pH值、酶添加量与超声时间、酶添加量与超声温度、pH值与超声时间、pH 值与超声温度和超声时间与超声温度的交互作用。因变量两两相互作用对SAPs 提取率的影响3D 响应面图均表现为光滑的弯曲面;四因素等高线图均表现为椭圆形状,表明相应变量之间的相互作用非常重要[28]。

图3 各因素交互作用对柳茶多糖提取率的影响的响应面图和等高线图Fig.3 Response and contour diagram of the effects of interactions of various factors on the extraction rate of SAPs
总之,优化的参数是酶质量分数为2%,pH 值为4.0,超声时间为2.0 h,超声温度为80 ℃。此时,SAPs提取率为10.95%,多糖含量为71.82%。较石磊[29]报道水提SAPs 的多糖含量为17%左右,说明PEG-UAEE提取存在较明显的提高效果,表明该优化提取条件是可供参考的。
2.4 相对分子质量分析
采用HPGPC 法测定超滤纯化后SAPs 的分子质量分布如表5 所示,根据标准洗脱曲线方程计算出SAPs分子量为1.5×103u,且信号峰呈现单一对称;此外,Mw/Mn值接近1 并低于已发表其他多糖的相对分子量[30,31],表明纯化后SAPs 的分子量分布均匀且单一。
2.5 单糖组成分析
2.5.1 薄层层析
如图4,样品水解完全与1 木糖、2 葡萄糖、3 阿拉伯糖、4 果糖、5 甘露糖、6 半乳糖、7 鼠李糖均呈现均一斑点,进一步可看出样品与葡萄糖、阿拉伯糖、甘露糖和半乳糖的位置接近。为进一步确定样品的单糖组分进行HPLC 分析。
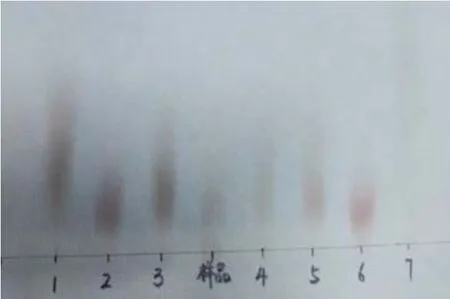
图4 多糖酸解后的薄层层析图Fig.4 TLC of the polysaccharide after acid degradation
2.5.2 HPLC 法分析单糖组成
根据HPLC-PMP 分析,7 种单糖标准品、SAPs 的色谱峰及出峰时间如图5 所示,对比5 种单糖标准品的保留时间,得出SAPs 中较明显的5 种单糖及占摩尔百分比分别为:半乳糖(13.34%)、葡萄糖(46.76%)、鼠李糖(8.78%)、果糖(2.89%)和阿拉伯糖(11.79%),纯化后SAPs 单糖含量进一步证明SAPs 是以半乳糖和葡萄糖为主的酸性杂多糖[32]。

图5 混合对照品(a)及柳茶单糖组成(b)HPLC 图Fig.5 HPLC chromatogram of monosaccharides of reference substances solution (a) and monosaccharides composition of SAPs (b)
2.6 红外光谱分析
图6 是SAPs 的红外光谱图,在3 366.11 cm-1处有很强的宽峰,为O-H 的伸缩振动吸收峰;2 879.75 cm-1附近为C-H 键伸缩振动引起的峰;这两组吸收峰为糖类的特征吸收峰,证明该提取物为多糖[33]。1 732.76 cm-1是糖醛酸中羰基C=O 振动,这表明柳茶多糖含有糖醛酸;1 611.16 cm-1处吸收峰是由乙酰基的C=O 振动引起的;1 436.25 cm-1处出现的吸收峰表明多糖主链可能有糖醛酸的存在;1 101.45 cm-1是糖环中C-O-C 伸缩和变角振动引起的峰,也表明其含有吡喃糖环;947.92 cm-1附近有较强拉伸峰,表明该多糖存在β型糖苷键[34]。根据红外光谱图出现的峰值越强,表明多糖含量越高可推断,SAPs 是β型吡喃多糖类化合物。

图6 柳茶多糖的红外光谱图Fig.6 Infrared spectra of SAPs
2.7 NMR 光谱分析
多糖的1H NMR 谱图中δ4.5~5.5 范围主要为多糖的糖苷键区域,该区域出现的质子信号表明了多糖的单糖种类数。其中,δ5.0 是区分吡喃糖构型的质子信号的临界值,当第一个碳的质子位移大于δ5.0 时,表明多糖为α-糖苷键否则为β-糖苷键[32]。纯化SAPs 的1H NMR 光谱如图7 所示。从图中可以看出,SAPs 在δ4.5~5.5 范围内有5 个质子信号峰,这与HPLC-PMP 结果一致,且第一个碳的质子位移出现在δ4.2~4.4,表明纯化SAPs 中存在β-糖苷键,这与FT-IR 分析结果一致。
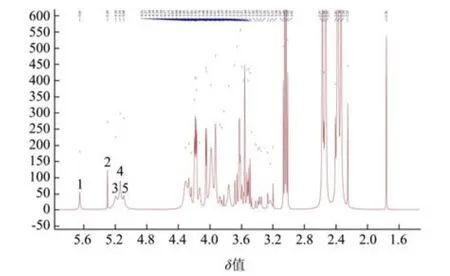
图7 柳茶多糖的1H NMR 图Fig.7 1H NMR spectra of SAPs
2.8 扫描电镜分析
SAPs 的微观扫描电镜结构如图8 所示,经观察可发现多糖主要由不规则碎片结构组成,伴有较多的细小不规则颗粒且表面较粗糙,结构致密且贴合度较高。由此可证明SAPs 具有明显的非晶态结构并较完整的结构形态。
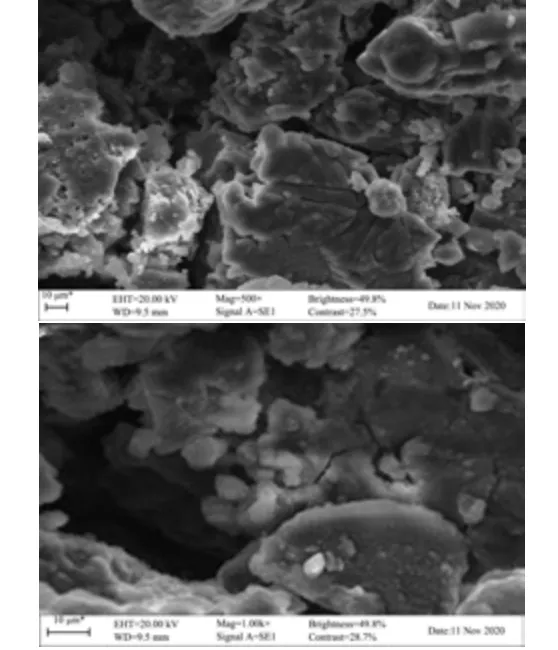
图8 柳茶多糖扫描电镜图Fig.8 Scanning electron microscopy of SAPs
2.9 抗氧化活性分析
DPPH 是一种稳定的有机氮合成自由基,与抗氧化剂提供的氢原子或电子结合而形成稳定的分子构型[35]。由图9a 可知,在0.0~1.0 mg/mL 范围内,SAPs 和Vc的DPPH·清除能力表现出明显的量效关系,随着质量浓度的增大而逐渐增大。DPPH·清除能力在SAPs 和Vc 质量浓度为0.0~0.2 mg/mL 时均呈明显上升,当多糖质量浓度为1.0 mg/mL 时,清除能力达到最大值80.33%,半抑制浓度IC50为0.42 mg/mL;此时Vc 清除能力也达到最大值95.12%,半抑制浓度IC50为0.21 mg/mL。表明SAPs 可作为食品加工和保鲜过程中的天然抗氧化剂。据相关研究表明,多糖的抗氧化活性与物质的分子量、硫酸化程度和糖苷键等存在关系,可通过超声波处理以降低分子量从而提高多糖物质的生物活性。
·OH 极易被氧化为各种有机物或无机物,在机体内可快速造成组织脂质过氧化、核酸断裂和蛋白质分解,从而造成导致组织损伤或细胞功能丧失[36]。由图9b 所示,SAPs 和Vc 对·OH 清除能力均表现出浓度依赖性,在0.0~1.0 mg/mL 范围内随着质量浓度的增大而逐渐增大。质量浓度达到0.4 mg/mL 后,SAPs 对·OH自由基清除能力趋势增长较平缓,当质量浓度达到1 mg/mL 时,多糖对·OH 清除能力达到最大值38.40%,半抑制浓度IC50为2.08 mg/mL;此时,Vc 对·OH 清除能力也达到最高值70.54%,半抑制浓度IC50为0.51 mg/mL。Wu 等[37]研究紫菜多糖含有α型和β型糖苷键结构,且清除·OH(22.84%,2 mg/mL)效率与低浓度SAPs 抑制·OH 接近。
·O2-是由机体内黄嘌呤脱氢酶、纤维二糖氧化酶和醛氧化酶等少数酶作用引起产生的重要自由基[38]。如图9c 所示,对·O2-清除能力在质量浓度为0.0~1.0 mg/mL范围内以明显的浓度依赖性方式增加。SAPs 质量浓度达到1.0 mg/mL 时,对·O2-清除能力达到最大值85.20%,半抑制浓度IC50=0.28 mg/mL;相同质量浓度下,Vc 清除·O2-能力高达102.31%,半抑制浓度IC50=0.20 mg/mL。Ma 等[39]研究发现,当天花粉蛋白多糖浓度达到1.28 mg/mL 时表现出最大·O2-清除活性为35.33%。研究表明,·O2-清除作用机制可能是由于醛类或酮类多糖中存在亲电基团。
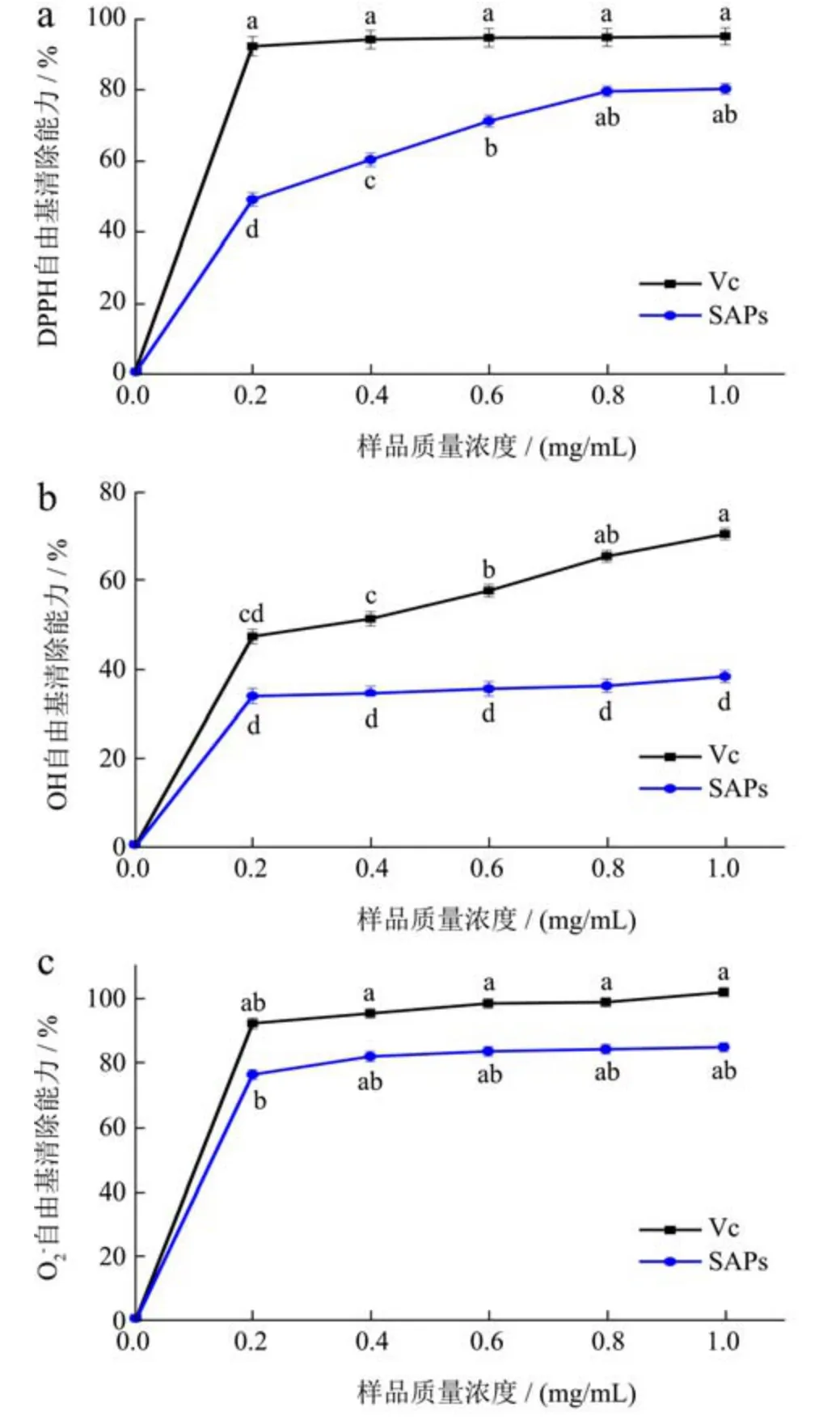
图9 柳茶多糖抗氧化活性Fig.9 Antioxidative activity of SAPs
综上可见,在0.0~1.0 mg/mL 质量浓度范围内,SAPs 和Vc 对·O2-清除能力均强于对DPPH·清除能力和·OH 清除能力,且清除活性较Vc 差距较小。
3 结论
以藏柳茶为原料,以多糖提取得率为评价指标,通过响应面试验优化PEG-UAEE 提取SAPs 最佳工艺为:PEG-400 浓度为30%,复合酶添加量2%,pH 值4.0,超声功率80%,超声时间2.0 h,超声温度80 ℃条件得出多糖提取率为10.95%,多糖含量为21.82%。由半乳糖、葡萄糖、鼠李糖、果糖和阿拉伯糖组成的酸性杂多糖,摩尔比为13.34:46.76:8.78:2.89:5.34:11.79;多糖具有完整明显的非晶态结构,β-糖苷键的吡喃型多糖;随着多糖含量的提高,其自由基清除能力剂量效应关系,DPPH·清除能力、·OH 清除能力和·O2-清除能力的IC50值分别为0.42、2.08、0.28 mg/mL。本研究可为藏药柳茶的进一步开发和利用提供参考。
