基于通用DNA条形码序列的黄精属药用植物分子鉴定
2023-01-10张明英李依民程文萍颜永刚胡锦航
张明英,李依民,程文萍,高 静,颜永刚,杨 琳,胡锦航,张 岗*
基于通用DNA条形码序列的黄精属药用植物分子鉴定
张明英1, 3,李依民1,程文萍1,高 静1,颜永刚1,杨 琳1,胡锦航2*,张 岗1, 3*
1. 陕西中医药大学药学院陕西省秦岭中草药应用开发工程技术研究中心,陕西 西安 712046 2. 陕西中医药大学陕西省中药资源产业化协同创新中心,陕西 咸阳 712083 3. 陕西中医药大学陕西省中医药管理局“秦药”研发重点实验室,陕西 西安 712046
分析4个通用植物DNA条形码序列(H-A、K、L和ITS2)及其组合对黄精属药用植物的物种鉴定分辨率,挖掘适用于黄精属种间鉴定的高分辨率分子标记。以《中国药典》2020年版中收录的黄精属药用植物黄精、滇黄精、多花黄精、玉竹及其地方常见同属替代品、混伪品共12种79个野生个体为对象,将4个通用DNA条形码序列独立、联合分析,评估其种间、种内变异情况,并分别基于建树法(tree-based method)和PWG距离法(PWG-distance method)评估不同条形码及其组合的物种鉴定分辨率。ITS2序列扩增成功率低,H-A、K、L序列的引物在黄精属植物中通用性较好;3组叶绿体序列的种间变异依次为K>H-A>L,种内变异差异不显著,种间、种内遗传距离无明显的Barcoding gap;各条形码独立及联合分析的物种鉴定分辨率普遍偏低,其中,组合条形码H-A+K+L在建树法分析中的分辨率最高,为25%,H-A+L在距离法分析中的分辨率最高,为50%。4个通用DNA条形码序列及其组合都并非黄精属药用植物不同种间有效区分鉴定的理想分子标记,但多序列联合分析能在一定程度上提高物种鉴定成功率。
黄精属;DNA条形码;药用植物;物种鉴定;黄精;滇黄精;多花黄精;玉竹
黄精属Mill.隶属于天门冬科(Asparagaceae)是一个具有重要药用和经济价值的草本植物类群。中国分布有黄精属植物39种[1],其中31种可入药使用[2],是中药材的重要来源。《中国药典》2020年版收载的大宗中药材黄精和玉竹均来源于黄精属药用植物。其中,黄精来源于滇黄精Coll. et Hemsl.、黄精Red.或多花黄精Hua的干燥根茎,具补气养阴、健脾、润肺、益肾之功效;玉竹则来源于玉竹(Mill.) Druce的干燥根茎,可养阴润燥、生津止渴,在《神农本草经》中曾被列为上品,中医临床应用广泛。此外,黄精属植物含多糖、甾体皂苷、黄酮、生物碱等多种活性成分,具有抗菌、抗氧化、抗肿瘤、降血糖、调血脂、调节免疫等药理作用[2-5],在药物开发研究中占据重要地位。
黄精属也是一个物种分类鉴定研究中的“困难”类群。自1754年Miller建立黄精属以来,其属下物种划分鉴定与种间系统发育关系问题长期受到关注。由于该属植物具有较高的形态多样性,自然野生状态下,不同种间、同种不同居群个体间的形态特征常存在过渡类型,且种间地理分布交叉重叠,导致依据形态性状不易对不同物种进行准确区分鉴定[6-10]。目前,黄精属种间分类鉴定问题仍有待澄清。此外,由于多以根状茎入药,生药性状相似,若经加工后,更是难以对其物种来源进行准确鉴别[7,11]。在我国不同地区,卷叶黄精(Wall.) Royle、湖北黄精Pamp.、长梗黄精Merr. ex C. Jeffrey et McEwan、轮叶黄精(L.) All.、新疆黄精(Ledeb.) Kunth等多种非正品基原代用、混用、误用,甚至伪品入药现象普遍存在[2, 7, 11],给该属植物来源中药材的用药安全和临床疗效带来隐患。
DNA条形码技术从分子遗传学角度为药用植物及中药材的客观准确鉴定提供了一条有效途径[12]。国际生命条形码联盟植物工作组(CBOL Plant Working Group)通过对7个叶绿体片段(L、K、C1、B、H-A、F-H和K-I)通用性(包括扩增成功率、测序成功率、序列质量)和物种鉴定分辨率的综合分析评估,推荐L+K组合可作为陆地植物鉴定的核心DNA条形码[13],并建议将ITS(包括ITS2)和HA作为辅助条形码。中国植物条形码研究团队(China Plant BOL Group)基于对种子植物75科141属1757种共6 286个代表个体的分析结果提出将ITS(或ITS2)纳入种子植物鉴定的核心条形码[14]。陈士林及其团队在药用植物及中药材研究中建立了以ITS2序列为主、HA序列为辅的DNA条形码分子鉴定体系[15]。这些条形码已被广泛用于药用植物类群如柴胡属L.[16]、五味子科(Schisandraceae)[17]、乌头属L.[18]、商陆属L.[19]等及其来源的中药材、饮片等的分子鉴定研究[20-22]。
目前,黄精属DNA条形码种间分子鉴定研究相对较少,杨培等[7]利用上述条码对黄精属8种44份样品的分子鉴定研究发现,ITS和ITS2序列扩增成功率低,HA、K及L序列的种间、种内变异均较小,物种分辨率不足。Jiao等[23]利用ITS2和HA对39份不同地区来源中药材黄精及其混伪品的鉴定分析表明,ITS2序列未能被成功扩增,HA序列虽能够将中药材黄精的基原物种与同属其他物种区分开来,但由于较低的遗传变异,对黄精属种间鉴别的分辨率依然有限。
本实验在前人研究基础上,以《中国药典》2020年版中收录的中药材黄精、玉竹基原植物及其在不同地区常见同属替代品、伪品来源植物为研究对象,从物种和种内个体水平增加取样量,利用上述4个DNA条形码序列(HA、K、L和ITS2)分别进行独立和联合分析,基于建树法(tree-based method)和PWG距离法(plant working group distance,PWG-distance method)开展分子鉴定研究,旨在评估各条码及其不同组合的物种鉴定分辨率,筛选适用于黄精属药用植物种间有效鉴定的分子标记,为该属植物来源中药材的准确鉴定、用药安全和药用植物资源保护及开发利用提供理论依据。
1 材料
以《中国药典》2020版中收录的黄精属药用植物黄精、滇黄精、多花黄精和玉竹,及文献资料中记载的其同属常见替代品、伪品共12个物种为研究对象,采集野外自然生长状态下的健康叶片,每种至少采集3个来自不同分布地点或居群的代表个体,硅胶快速脱水干燥保存,用于DNA提取,同时采集凭证标本。本研究共采集到57个代表个体,标本经陕西中医药大学标本馆王继涛高级实验师鉴定,保存于陕西中医药大学中药标本馆。另有22个代表个体的序列数据由中国西南野生生物种质资源库提供,即总共79个代表个体(表1)。
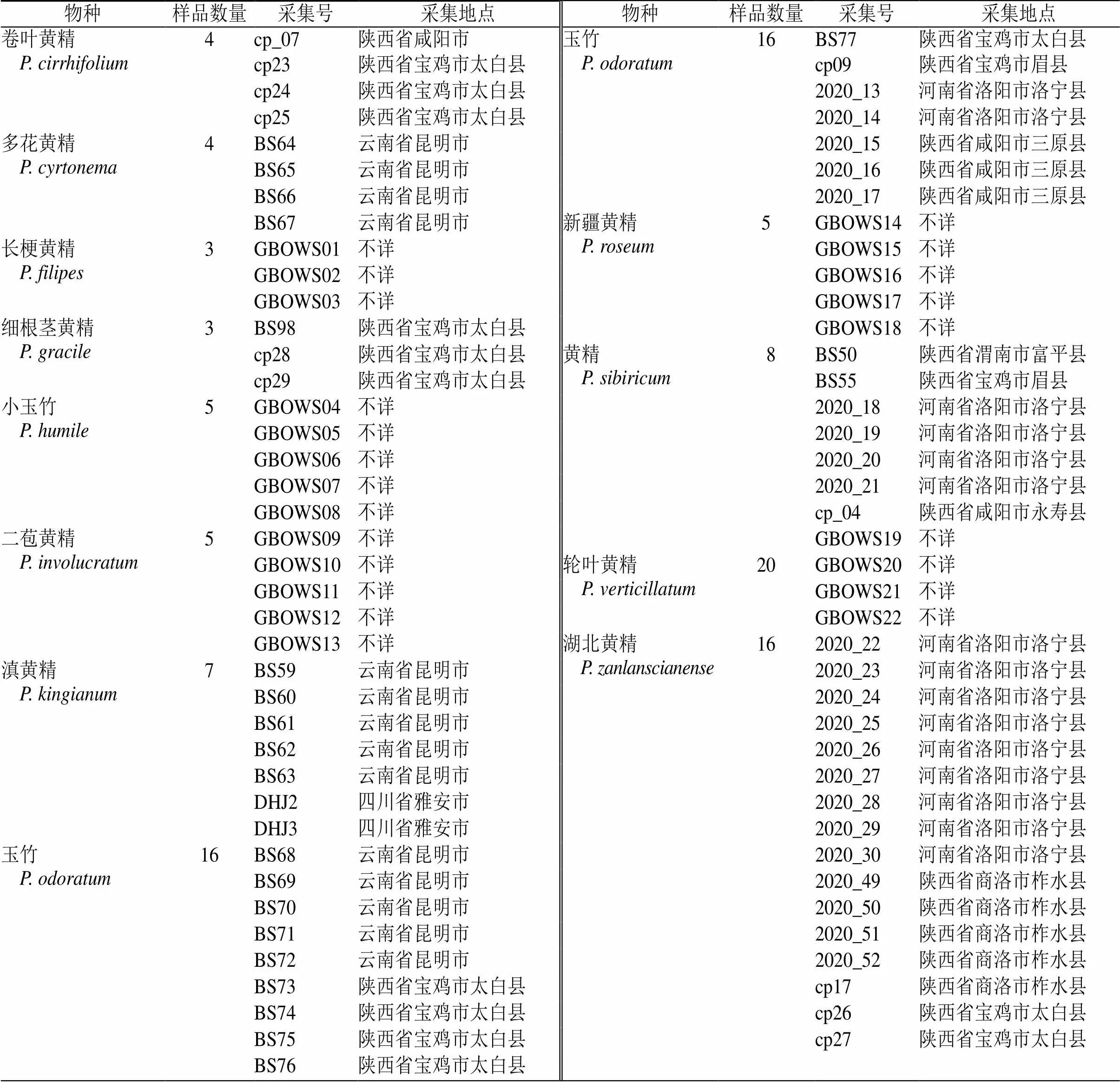
表1 材料信息
2 方法
2.1 基因组DNA提取、PCR扩增和测序
采用改良的CTAB法提取基因组总DNA,TE缓冲液溶解,得到的DNA溶液用1%的琼脂糖凝胶电泳和NanoDrop 2000分光光度计(Thermo Fisher Scientific,Delaware,美国)检测质量与浓度。PCR扩增反应在GeneAmp PCR System 9700 thermal cycle(Applied Biosystems, Foster City,CA,美国)上进行。引物信息见表2。PCR反应体系均为50 µL,包括2×Taq Plus PCR MasterMix 25 µL,10 µmol/L的正、反向引物各2 µL,模板DNA 10 µL,ddH2O补齐。根据文献报道结合前期预实验结果,最终选用的PCR扩增程序为:94 ℃预变性4 min,94 ℃变性30~45 s,54.5~57.0 ℃退火30 s(各引物退火温度详见表2),72 ℃延伸60 s,重复35个循环,72 ℃延伸10 min,结束后4 ℃保存。取5 µL扩增产物,用1%的琼脂糖凝胶电泳检测。成功扩增的PCR产物进行双向测序。PCR引物合成和测序均由生工生物工程(上海)股份有限公司西安分部完成。
2.2 序列拼接、比对及特征分析
测序原始结果利用Sequencher软件进行序列拼接,并根据测序峰图对碱基进行校正,去除引物区及两端低质量序列,将得到的所有序列分别在NCBI数据库中进行Blastn(https://blast.ncbi.nlm. nih.gov/Blast.cgi)比对,验证序列的正确性。对拼接得到的ITS序列采用基于隐马尔可夫模型的HMMer注释方法去除两端5.8 S和26 S区,获得ITS2间隔区序列[29]。由于ITS2序列扩增成功率较低,最终获得的序列数量低于实验个体总数的50%,因而未将其纳入后续分析。

表2 PCR扩增引物信息
最终得到的3组叶绿体序列(HA、K、L)分别构建多序列矩阵,利用MAFFT完成序列比对,并在Geneious软件中进行必要的人工检查校正。将3组叶绿体序列分别独立和两两、3个联合进行分析,即HA、K、L、HA+K、HA+L、K+L和HA+K+L,共7组条形码序列。对于3组独立条形码序列,利用MEGA软件统计变异位点(variable sites)和简约信息位点(parsimony informative sites),并计算种间、种内K2P(kimura 2-parameter distance)遗传距离,利用IMB SPSS Statistics 25进行Wilcoxon符号秩检验,分析各组序列的种间、种内遗传变异差异显著性。联合分析,即将3组独立序列按不同组合分别进行串联合并,若某个体有序列缺失情况(如2条序列联合时,某个体只有1条序列;或3条序列联合时,某个体只有1条或2条序列),则将该个体在分析中去除。
2.3 物种鉴定分析
利用MEGA软件分别计算上述7组条形码序列的种间、种内K2P距离,利用TaxonDNA结合统计软件对种间、种内K2P距离分布频度进行统计并绘图,评估种间和种内遗传距离间是否存在“Barcoding gap”。
采用建树法和PWG距离法2种分析方法评估7组条形码的物种鉴定分辨率。对于建树法,利用MEGA软件基于K2P距离和配对删除(pair-deletion)模型,构建邻接系统发育树(neighbor-joining tree,NJ tree),系统发育树各分支节点的靴带支持率(bootstrap values,BS)通过进行1000次自展重复分析获得。当同一物种的所有个体在系统树上聚为一个单系,且支持率高于50%,则视为该物种鉴定成功[14, 30]。对于PWG距离法,利用MEGA软件分别计算各物种的种间、种内遗传距离,当某一物种与其他物种间的最小遗传距离大于该物种种内个体间的最大遗传距离时,则表示该物种鉴定成功[13]。
3 结果与分析
3.1 PCR扩增、测序结果及序列特征
PCR扩增、测序结果及各序列相关信息见表3。3组叶绿体条形码序列中,HA的扩增和测序成功率均达到100%;L的扩增成功率为100%,有2个个体测序失败,即测序成功率为97.5%;K的扩增成功率为88.6%,扩增成功的个体全部成功测序。ITS2仅41个样品扩增成功,即扩增成功率为51.9%,其中2个个体测序失败,最终仅得到39条序列。扩增失败的样品经调整扩增反应条件(退火温度、酶及引物、模板DNA量等)后电泳,仍未检测到明亮、单一的扩增产物条带。
3组叶绿体序列比对后的长度为499~751 bp,变异位点和信息位点含量分别为1.5%~4.53%和1.2%~1.86%,其中K序列长度最长,且变异和信息位点含量均为最高。玉竹有9个个体(BS73~75、BS77、2020_13~17)的HA序列在60~67 bp位置发生8 bp的倒位(GTTTTCAT→ ATGAAAAC)。
3.2 Wilcoxon符号秩检验
Wilcoxon符号秩检验结果见表4,3组叶绿体序列的种间变异由大到小依次为K>HA>L,而种内差异不显著,即K=HA=L。
3.3 Barcoding gap检验
Barcoding gap检验结果见图1,所有7组条形码序列的种间、种内遗传距离均存在一定程度重叠,即没有明显的Barcoding gap存在。种内距离分布较为集中,主要在0~0.004,种间遗传距离相对离散,主要在0.003~0.01,序列联合分析的种间、种内遗传距离重叠程度小于独立分析。

表3 PCR扩增、测序结果及比对后各组序列特征

表4 TrnH-psbA、matK及rbcL序列的种间、种内变异Wilcoxon符号秩检验
3.4 物种鉴定分辨率
2种分析方法所得的各条形码的物种鉴定分辨率见表5。建树法分析结果中,独立及联合分析的分辨率为8.33%~25.00%,成功鉴定1~3个物种。其中,HA和K序列分别以74%和85%的支持率支持黄精种内所有个体聚为单系,与其他物种区分开来,L序列分析结果支持长梗黄精所有个体单独构成一个单系,与其他物种相区别,支持率为94%。联合分析中,2个条形码组合HA+K、HA+L及K+L成功鉴别的物种数均为2个,依次分别为滇黄精(支持率为51%)和黄精(97%)、长梗黄精(95%)和滇黄精(80%)及长梗黄精(57%)和黄精(88%),鉴定成功率为16.66%。而长梗黄精、滇黄精和黄精3个物种同时被HA+K+L组合成功鉴定,支持率依次为94%、69%和96%(图2)。
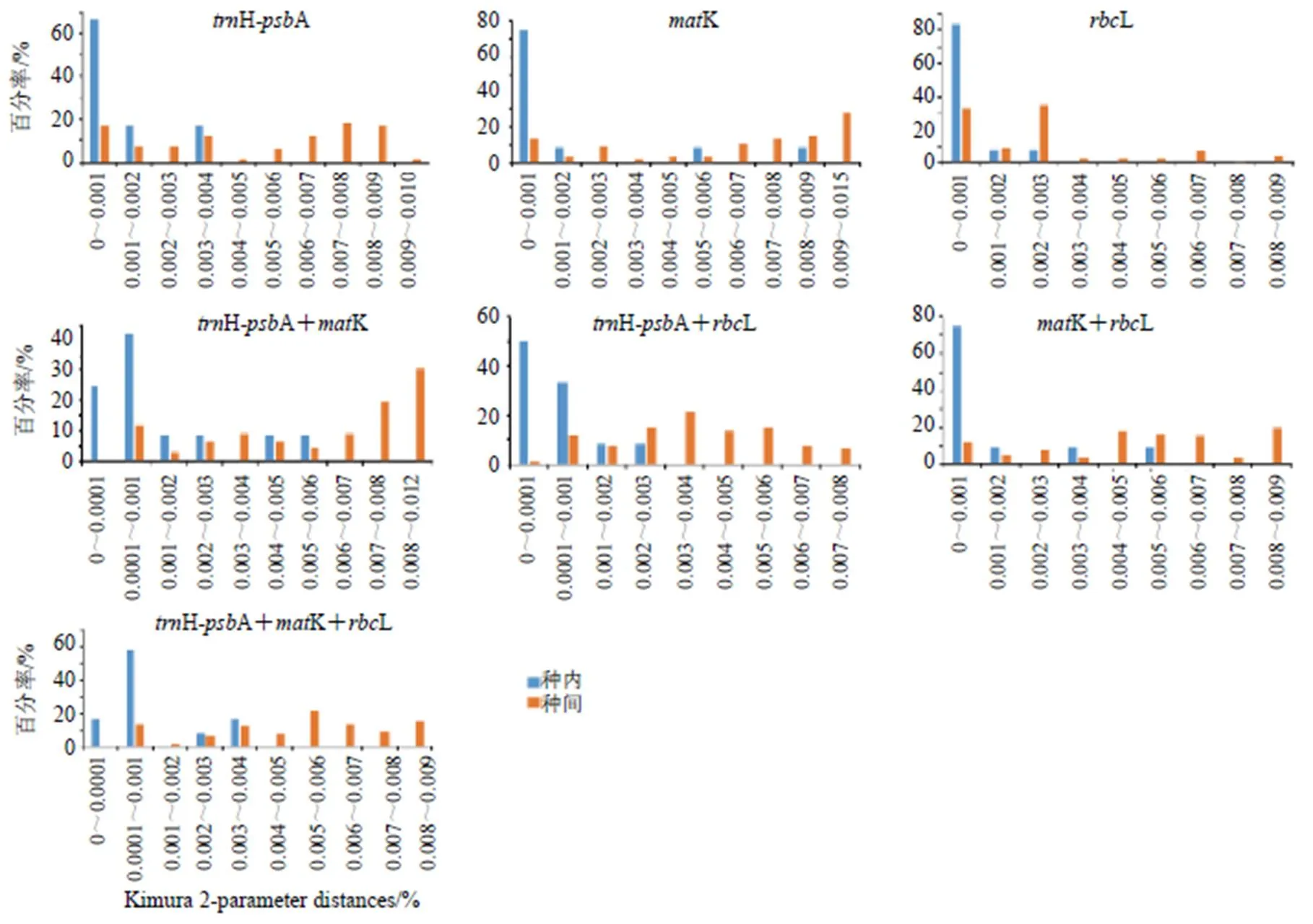
图1 各条形码序列及其不同组合的种间、种内遗传距离分布图

表5 基于建树法和PWG距离法分析的各条形码序列及其不同组合的物种鉴定分辨率

图2 TrnH-psbA、matK和rbcL序列联合分析构建的邻接系统发育树
PWG距离法分析的物种鉴定分辨率为8.33%~50%,成功鉴定的物种数为1~6个。3个条形码独立分析的物种鉴定成功率依次为41.67%(K)、16.66%(L)和8.33%(HA)。组合条形码中,HA+L共包含12种77个个体,其中,多花黄精、长梗黄精、二苞黄精、滇黄精、新疆黄精和轮叶黄精6个物种的种内最大遗传距离均小于其与其他物种间的最小遗传距离而被成功鉴定,其余条形码组合成功鉴定的物种数量均为5个(41.67%)。
4 讨论
通用性高、测序质量好、物种分辨率高是评价DNA条形码序列的重要指标[13-14]。本研究结果中,3个叶绿体条形码序列HA、K、L的PCR扩增成功率分别为100%、88.6%和100%,除L有2个个体测序失败外,其余扩增成功的个体全部成功测序,说明这3条序列的引物在黄精属植物中通用性较好。
Chen等[15]对药用植物(包括藻类、真菌及高等植物)753属4800种6600个代表个体的分析结果表明,ITS2序列在物种水平的鉴定率高达92.7%,因此推荐将ITS2作为药用植物分子鉴定的标准DNA条形码序列。然而,ITS/ITS2在一些类群中仍然存在难以成功扩增和测序的问题[31],黄精属便是这样一个类群。本研究结果显示,ITS2的PCR扩增成功率远低于其他3个叶绿体序列,仅为51.9%,调整扩增反应条件后仍未得到改善,79个个体最终只成功测序获得39条序列。Jiao等[23]对不同产地来源中药材黄精样品及其混伪品的分子鉴定研究也发现ITS2序列不能被特异性扩增成功,杨培等[7]对黄精属药用植物的分子鉴定研究亦曾得出相似的结论。PCR扩增结果主要受到引物通用性和实验材料个体序列变异的影响。研究表明,利用基因组浅层测序(genome skimming)方法可以有效避免这些问题而获取核基因组中的ITS序列[30, 32],这将是解决黄精属分子鉴定及相关研究中ITS2序列获取问题的一个新选择。
理想的DNA条形码序列种内遗传距离应明显小于种间,即具有明显的Barcoding gap,而本研究所有7组条形码序列的种间、种内遗传距离均存在一定程度重叠,无明显的Barcoding gap。此外,在建树法分析结果中,HA+K+L组合的物种鉴定成功率最高,为25%,即所有12个物种中仅3个物种被同时成功鉴定;HA+L组合在距离法中的物种分辨率最高,为50%,仅6个物种被同时成功鉴定。说明叶绿体序列HA、K、L及其组合都并非黄精属药用植物不同种间有效区分鉴定的理想分子标记。尽管如此,从本研究两种分析结果可以明显看出,随着序列数量的增加,物种鉴定分辨率也相应提高。建树法分析结果中,3个叶绿体序列独立分析的物种鉴定分辨率均为8.33%(1/12),两两组合(HA+K、HA+L、K+L)后分辨率提高为16.66%(2/12),HA+K+L三者联合分析的物种鉴定分辨率提高至25%(3/12)。距离法分析结果中K独立分析的物种分辨率最高,为41.67%,HA和L分别为16.66%和8.33%,而序列组合HA+L的物种分辨率提高至50%。说明多个条形码序列联合分析能够提供更多的物种演化信息位点,在一定程度上提高物种鉴定分辨率,这也与五味子科[17]、地黄属Libosch. ex Fisch. & C. A. Mey.[33]等药用植物类群的DNA条形码分子鉴定研究结果一致。
被子植物叶绿体基因组大小一般在115~165 kb,编码约110~130个基因,由于其序列进化速率适中,极少发生重组、基因含量和顺序高度保守,而所包含的物种演化信息量远大于单一或多个普通的DNA条形码序列,作为“超级条形码(ultra-barcode),近年来,在药用植物分子鉴定研究中广泛应用。如Zhang等[34]利用叶绿体全基因组序列将通用DNA条形码K+L、ITS+H-A联合分析未能区分鉴定的菊科Compositae紫锥菊属Moench 9个物种有效鉴别开来,为具有药用价值的紫锥菊(L.) Moench、狭叶紫锥菊DC.和白色紫锥菊(Nutt.) Nutt.及其混伪品的准确鉴定提供了可靠依据。Zhu等[35]利用叶绿体全基因组序列将ITS2及K+L均不能有效区分鉴定的名贵中药材铁皮石斛Kimura et Migo与其同属5个近缘物种黄石斛Makino、始兴石斛Z. L. Chen,S. J. Zeng & J. Duan、曲茎石斛Z. H. Tsi, S. C. Sun & L. G. Xu、滇桂石斛W. W. Sm.和钩状石斛Wall. ex Lindl.以≥99%的分辨率成功鉴定,为铁皮石斛的入药安全和有效性提供了重要保障。Yin等[36]的研究结果也表明利用叶绿体全基因组序列可以将蔷薇属L.药用植物金樱子Michx.、玫瑰Thunb.、犬蔷薇L.以及月季花Jacq.与同属其他物种有效区分鉴别。此外,Flodena等[37]对黄精属19个代表物种的分子系统发育分析也进一步表明,相比于单个或少数几个基因序列的分析结果,利用叶绿体基因组中的全部蛋白编码基因序列联合分析所得的系统发育树分辨率明显提高。因此,叶绿体全基因序列将有可能是解决黄精属药用植物种间鉴定困难问题的一条有效途径,有待后续进一步研究验证。
利益冲突 所有作者均声明不存在利益冲突
[1] Chen X Q, Tamura M N.24 [M]. Beijing: Science Press/St. Louis: Missouri Botanical Garden Press, 2000: 225-235.
[2] 艾铁民. 中国药用植物志(第十一卷) [M]. 北京: 北京大学医学出版社, 2014: 277-306.
[3] 张娇, 王元忠, 杨维泽, 等. 黄精属植物化学成分及药理活性研究进展 [J]. 中国中药杂志, 2019, 44(10): 1989-2008.
[4] 姜程曦, 张铁军, 陈常青, 等. 黄精的研究进展及其质量标志物的预测分析 [J]. 中草药, 2017, 48(1): 1-16.
[5] Zhao X Y, Li J. Chemical constituents of the genusand their role in medicinal treatment [J]., 2015, 10(4): 683-688.
[6] Meng Y, Nie Z L, Deng T,. Phylogenetics and evolution of phyllotaxy in the Solomon's seal genus(Asparagaceae: Polygonateae) [J]., 2014, 176(4): 435-451.
[7] 杨培, 周红, 辛天怡, 等. 黄精属药用植物DNA条形码鉴定研究 [J]. 世界中医药, 2015, 10(8): 1173-1176.
[8] Zhao L H, Zhou S D, He X J. A phylogenetic study of Chinese(Polygonateae, Asparagaceae) [J]., 2019, 37(2): njb.02019.
[9] Zhao L H, Zhou S D, He X J,. A cytotaxonomic analysis of Chinese(Asparagaceae) species [J]., 2014, 32(4): 441-451.
[10] Wang J J, Yang Y P, Sun H,. The biogeographic south-north divide of(Asparagaceae tribe polygonateae) within eastern Asia and its recent dispersals in the Northern Hemisphere [J]., 2016, 11(11): e0166134.
[11] 林琳, 林寿全. 黄精属药用植物聚类分析 [J]. 中药材, 1994, 17(6): 12-18, 54.
[12] 陈士林, 庞晓慧, 罗焜, 等. 生物资源的DNA条形码技术 [J]. 生命科学, 2013, 25(5): 451-459.
[13] CBOL Plant Working Group. A DNA barcode for land plants [J]., 2009, 106(31): 12794-12797.
[14] Li D Z, Gao L M. Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants [J]., 2011, 108(49): 19641-19646.
[15] Chen S L, Yao H, Han J P,. Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species [J]., 2010, 5(1): e8613.
[16] Chao Z, Zeng W P, Liao J,. DNA barcoding Chinese medicinal[J]., 2014, 21(13): 1767-1773.
[17] Zhang J, Chen M, Dong X Y,. Evaluation of four commonly used DNA barcoding Loci for Chinese medicinal plants of the family Schisandraceae [J]., 2015, 10(5): e0125574.
[18] 任瑶瑶, 蔡子君, 赵梅宇, 等. 基于ITS2条形码的乌头属藏药植物鉴别 [J]. 中草药, 2018, 49(19): 4614-4620.
[19] 吕瑞华, 冯昭, 马添翼, 等. 陕西关中野生商陆资源的ITS2和psbA-trnH条形码序列研究 [J]. 药学学报, 2020, 55(8): 1951-1956.
[20] 袁伯川, 李文东, 马永生, 等. 柴胡属药用植物的分子鉴定及市售柴胡药材的质量调查 [J]. 药学学报, 2017, 52(1): 162-171.
[21] 辛天怡, 娄千, 郝利军, 等. 市售中药饮片DNA条形码鉴定研究 [J]. 药学学报, 2021, 56(3): 879-889.
[22] 熊瑶, 金晨, 王晓云, 等. 鸡血藤及其混伪品的DNA条形码分子鉴定研究 [J]. 中草药, 2020, 51(12): 3274-3283.
[23] Jiao J, Huang W L, Bai Z Q,. DNA barcoding for the efficient and accurate identification of medicinal polygonati rhizoma in China [J]., 2018, 13(7): e0201015.
[24] White T J, Bruns T, Lee S,. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics [A] //[M]. Amsterdam: Elsevier, 1990: 315-322.
[25] Tate J A, Simpson B B. Paraphyly of(Malvaceae) and diverse origins of the polyploid species [J]., 2003, 28(4): 723-737.
[26] Sang T, Crawford D, Stuessy T. Chloroplast DNA phylogeny, reticulate evolution, and biogeography of(Paeoniaceae) [J]., 1997, 84(8): 1120.
[27] 高连明, 刘杰, 蔡杰, 等. 关于植物DNA条形码研究技术规范 [J]. 植物分类与资源学报, 2012, 34(6): 592-606.
[28] Kress W J, Erickson D L. A two-locus global DNA barcode for land plants: The codingL gene complements the non-coding trnH-psbA spacer region [J]., 2007, 2(6): e508.
[29] Keller A, Schleicher T, Schultz J,. 5.8S-28S rRNA interaction and HMM-based ITS2 annotation [J]., 2009, 430(1/2): 50-57.
[30] Fu C N, Mo Z Q, Yang J B,. Testing genome skimming for species discrimination in the large and taxonomically difficult genus[J]., 2022, 22(1): 404-414.
[31] Hollingsworth P M. Refining the DNA barcode for land plants [J]., 2011, 108(49): 19451-19452.
[32] Hollingsworth P M, Li D Z, van der Bank M,. Telling plant species apart with DNA: From barcodes to genomes [J]., 2016, 371(1702): 20150338.
[33] 程芳婷, 李忠虎, 刘春艳, 等. 地黄属植物的DNA条形码研究 [J]. 植物科学学报, 2015, 33(1): 25-32.
[34] Zhang N, Erickson D L, Ramachandran P,. An analysis ofchloroplast genomes: Implications for future botanical identification [J]., 2017, 7(1): 216.
[35] Zhu S Y, Niu Z T, Xue Q Y,. Accurate authentication ofand its closely related species by comparative analysis of complete plastomes [J]., 2018, 8(6): 969-980.
[36] Yin X M, Liao B S, Guo S,. The chloroplasts genomic analyses ofand[J]., 2020, 15: 18.
[37] Flodena A, Schillingb E E. Using phylogenomics to reconstruct phylogenetic relationships within tribe Polygonateae (Asparagaceae), with a special focus on[J]., 2018, 129: 202-213.
Molecular authentication of medicinalSpecies utilizing the universal DNA barcode sequences
ZHANG Ming-ying1, 3, LI Yi-min1, CHENG Wen-ping1, GAO Jing1, YAN Yong-gang1, YANG Lin1, HU Jin-hang2, ZHANG Gang1, 3
1. Shaanxi Qinling Application Development and Engineering Center of Chinese Herbal Medicine, College of Pharmacy, Shaanxi University of Chinese Medicine, Xi’an 712046, China 2. Shaanxi Collaborative Innovation Center of Chinese Medicinal Resources Industrialization, Shaanxi University of Chinese Medicine, Xianyang 712083, China 3. Key Laboratory for Research of "Qin Medicine" of Shaanxi Administration of Traditional Chinese Medicine, Shaanxi University of Chinese Medicine, Xi’an 712046, China
To investigate the species discrimination power of the four universal plant DNA barcodes (H-A,K,L and ITS2) and corresponding multi-barcode combinations in, and to explore high-resolution molecular markers suitable for.Seventy-nine wild individuals from 12 species, representing all the four medicinal species of(,,,) included in the(2020 Edition) and their local commonly used substitutions and inauthentic adulterants, were sampled. The interspecific and intraspecific genetic variation were estimated, tree-based and PWG-distance methods were applied to evaluate the species discrimination efficiency of each barcode sequence and their combinations.The primers ofH-A,K andL all showed good universality while most of the individuals failed to obtain ITS2 sequence in PCR amplification. The interspecific genetic variation of the three chloroplast sequences wasK>H-A>L, while their intraspecific genetic difference was not significant, and no obvious Barcoding gap was detected. All these barcode sequences including their combinations only get limited species resolution. Among which, the combination ofH-A+K+L possessed the best species-resolving power of 25% in tree-based method,H-A+L showed the highest resolution degree of 50% in PWG-distance method.None of the four barcode sequences nor their combinations were ideal molecular markers to address the problems of medicinalspecies authentication. Nonetheless, as the number of sequence increases, the degree of species resolution improves.
Mill.; DNA barcode; medicinal plant; species discrimination;Red;Coll. et Hemsl.;Hua;L.
R286.12
A
0253 - 2670(2023)01 - 0235 - 10
10.7501/j.issn.0253-2670.2023.01.025
2022-06-06
国家自然科学基金项目(82003898);陕西省自然科学基础研究计划项目(2022JM-458);陕西中医药大学校级科研课题(2020GP34);陕西中医药大学博士科研启动经费(104080001);陕西中医药大学“秦药”品质评价及资源开发学科创新团队项目(2019-QN01)
张明英(1988—),女,讲师,博士,研究方向为分子生药学。E-mail: zhangmy@sntcm.edu.cn
通信作者:张 岗,教授,研究方向为药用植物生物技术与分子生物学。E-mail: jay_gumling2003@aliyun.com
胡锦航,讲师,博士,研究方向为抗肿瘤药物药理。E-mail: hujinhanghi@126.com
[责任编辑 时圣明]
