基于含时密度泛函理论的紫外光对太安的定性影响
2023-01-10张宝森张树海苟瑞君陈亚红朱双飞马坤
张宝森,张树海,苟瑞君,陈亚红,朱双飞,马坤
基于含时密度泛函理论的紫外光对太安的定性影响
张宝森1,张树海1,苟瑞君1,陈亚红1,朱双飞1,马坤2
(1.中北大学 环境与安全工程学院,太原 030051;2.陕西应用物理化学研究所,西安 710061)
研究太安炸药在紫外光作用下的稳定性退化机制。基于TDDFT(含时密度泛函)理论,在pbe1pbe/6-311G**水平下对太安分子50个激发态进行计算,依据计算结果,绘制吸收光谱,使用空穴–电子方法对最大吸收峰3个激发态(9、10和11)的激发特征进行分析,这3个激发态对最大吸收峰的总贡献率达97.31%。将此3个激发态定为研究对象,对太安分子被特定的紫外光激发至激发态后弱键的Mayer和Laplace键级进行分析,并基于IFCT(Interfragment Charge Transfer)方法对太安分子激发至激发态过程中的电子转移情况进行描述。太安紫外吸收光谱的最大吸收峰位置为186.6 nm,小于实验测试的吸收峰位置8.4 nm。对此吸收峰的强度贡献最大的3个激发态中,9态贡献最高,为48.27%,其余2个激发态10、11为简并态,贡献相同,为24.52%。通过空穴–电子的电荷转移分析结果可知,3个激发态均存在整体激发并带有局域电荷转移的特征。PETN分子在吸收特定波长(187.00、186.92 nm)紫外光并激发至激发态时,O—NO2键的Mayer与Laplace键级均有所降低。通过IFCT分析可知,引发键键级变化由O—NO2上的n→Pi*跃迁主导,这种效应会促使太安分子的稳定性降低。
太安;紫外光;含时密度泛函;吸收光谱;分子稳定性;激发态
季戊四醇四硝酸酯(太安,PETN)因其具有较高的化学稳定性和爆轰性能,被广泛用于雷管、导爆索以及传爆药的制造之中[1]。近年来,激光起爆技术以其安全、可靠、低成本和对静电不敏感等传统技术无法代替的特点在国内外火工品中得到广泛应用[2-5]。
1996年,Tarzhanov等[6]对1.06 μm激光起爆PETN的实验结果进行了分析,分析结果表明,1.06 μm激光作用下的PETN响应实验结果符合光热起爆理论。2008年,陕西应用物理化学研究所[7]对多种配合物起爆药、炸药的吸收光谱及其不同波长下的激光感度进行了测试,测试结果表明,可见紫外波段激光或将破坏化合物的弱键,并引发化学反应。同时,激光敏感药剂的感度选择性调控,可以通过化合物辐射跃迁的选择定律和弱化合键的分解机理来实现。2011年,Aluker等[8]对钕玻璃激光作用添加光散射剂后的PETN样品进行了研究,研究表明,MgO光散射剂的添加会显著提升样品对激光能量的转换概率。2013年,中国工程物理研究院化工材料研究所[9]采用实验结合量子化学计算的方法,对多种炸药的激光敏感波长进行了分析,其中PETN最弱化学键为O—NO2键,实验测得PETN最敏感的紫外激光波长值为195 nm。2014年,Tsyshevsky等[10]对PETN光学吸收光谱中的能带进行了分析,强度较高能带对应于一系列的单线态–单线态跃迁,而强度较低的能带归因于紧密结合的单线态和三线态激子,所有跃迁过程都位于PETN的O—NO2基团。2017年,Tarzhanov等[11]研究了Al粉与PETN混合物的激光起爆特性,研究表明,Al粉与PETN粉混合形成的沉淀物的起爆难度较高。2019年,Aduev等[12-13]对PETN/Al复合物的激光起爆机理进行了研究,结果表明,样品中Al的添加会降低样品激光起爆的临界能量密度,当复合物中含0.1%(质量分数)铝时,样品的临界能量密度最低。2020年,北京理工大学严楠团队[14]使用仿真模拟与实验相结合的方法,对半导体激光直接起爆炸药的可行性进行了研究,验证了半导体激光直接起爆炸药的可行性。
近红外激光起爆炸药,通过添加掺杂物的方法提升炸药的激光感度,以激光热效应完成起爆过程。可见紫外激光是通过选择性激发炸药分子的电子能级,降低化合物中弱化学键的稳定性,诱导化学反应的发生。因此,研究激光器紫外–可见波长与药剂吸收光谱之间的匹配技术是提高激光特征感度的有效途径之一[7]。本文将借助TDDFT理论,对炸药分子激发态进行计算,对比激发前后分子引发键强度,以研究紫外光作用下炸药的稳定性退化机制。在分子层面,研究PETN在紫外光下的定性响应将为特征感度寻找提供数据支撑,为含能材料在激光环境下的防护理论依据。
1 计算细节及结构来源
分子对光的吸收过程涉及振动态和电子态的改变,在实际的计算过程中,往往只考虑电子的势能面,而忽视核振动的量子效应,其目的是简化计算。本文计算的PETN分子结构来源于CCDC晶体数据库[15],使用Gaussian09[16]程序在pbe1pbe/6-311G**水平下对PETN分子结构进行优化,结果无虚频。借助TDDFT理论,在pbe1pbe/6-311G**水平下基于优化后结构(如图1所示),对PETN的50个激发态进行计算。使用Multiwfn[17]软件,借助展宽函数和FWHM(Full Width at Half Maximum)对PETN 50个激发态下的紫外吸收光谱进行绘制,并与实验测试的PETN吸收光谱最大吸收峰位置[9]进行对比。
为明确PETN分子的跃迁特征,分析了激发态对吸收峰的贡献值。使用空穴–电子[18]的方法对贡献最高的3个激发态的电子和空穴分布进行了绘制,空穴–电子分析方法已多次用于含能材料的电子激发态研究[19-20]。为研究PETN激发过程中的电荷转移情况,进一步使用IFCT[17]方法对PETN片段间电子转移量进行计算,并使用Gaussian09软件对3个激发态密度进行计算。通过Multiwfn软件生成自然轨道,以对不同态下O—NO2键的Mayer与Laplace键级进行计算。
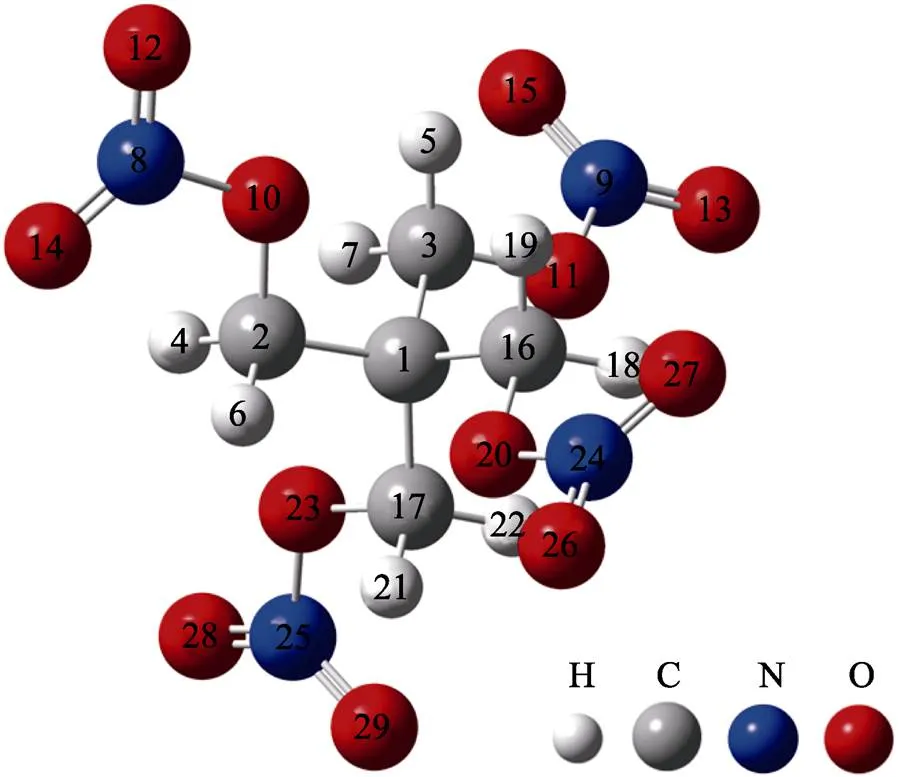
图1 PETN分子优化后结构及原子编号
2 结果及分析
2.1 PETN分子紫外吸收光谱与激发特征分析
使用Gaussian09软件,基于优化后的PETN基态结构,依据含时密度泛函理论,对其50个激发态进行了计算。借助Multiwfn,依据激发态计算结果,绘制PETN紫外吸收光谱(如图2所示),其中FWHM 参数设定为0.7 eV,计算水平为pbe1pbe/6-311G**。
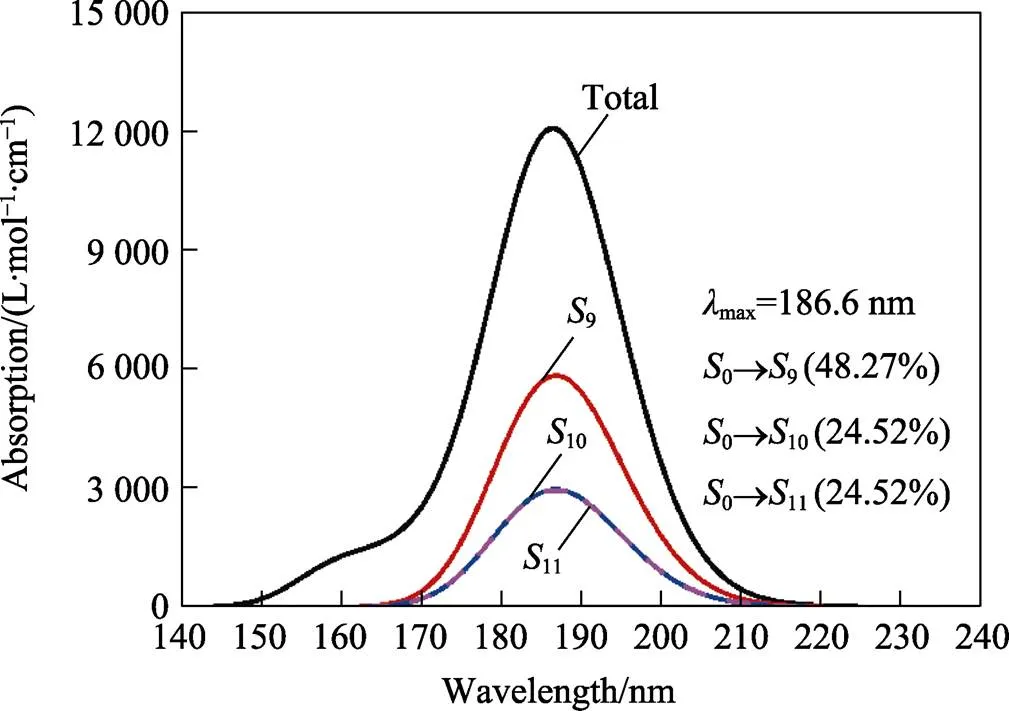
图2 基于TDDFT方法计算的PETN分子的紫外吸收光谱
由图2可知,PETN分子计算光谱的最大吸收峰位置为186.6 nm。在以往的研究中,实验测得PETN炸药在195 nm处[9]的吸光度最高,与本文所计算的紫外吸收光谱最大吸收峰位置仅相差8.4 nm。由此可知,本文计算的紫外吸收光谱与实验测试的吸收光谱符合较好。
PETN分子吸收光谱中,对最大吸收峰起到主要贡献的3个激发过程为0→9、0→10、0→11,贡献率分别为48.27%、24.52%、24.52%,振子强度分别为0.144、0.073、0.073。其中,振子强度表示分子吸收某一波长的光的概率[21]。在3种激发态中,10与11对波峰的贡献率相同,并且2个态的激发能相等,均为6.633 0 eV,由此可以确定10和11为简并态。
计算3个激发态的轨道跃迁贡献(大于5%),结果见表1。其中10与11态的2类轨道跃迁贡献率相近,2个激发态的激发能相等。LUMO+1与LUMO+2能量分别为–2.130 1、–2.130 0 eV,2个分子轨道能量数值上近似相等,由此可将这2轨道视为简并轨道。
表1 3个主要贡献的激发过程的主要轨道跃迁贡献率

Tab.1 Main orbital transition contribution rates in excitation processes of excited states S9, S11 and S10
空穴–电子方法适用于描述大部分体系的激发过程。为明确以上激发态的激发特征,本文将采用空穴–电子方法来分析PETN的电子激发特征。空穴–电子分析是基于空穴和电子的密度分布,“空穴”是指电子供体,是电子被激发和转移的地方;而“电子”表示电子受体。
表2中指数表示空穴和电子质心之间的距离;r指数表示电子和空穴的重叠程度;指数表示电子空穴的总体平均分布广度;指数衡量空穴和电子的分离程度;coul表示空穴与电子间的库仑吸引能;空穴离域指数(HDI)和电子离域指数(EDI)表示空穴或电子的分布均匀程度。由表2可知,3种激发态指数、r指数数值均较小,而指数大于0.25 nm。通过以上3种参数可以确定,3种激发过程具有整体激发特征。3种激发态的指数为负值,表示在电荷转移激发方向上,空穴与电子的分布未发生显著的分离。HDI与EDI数值相近,表示空穴和电子的分布均匀程度类似,这与以上分析相符合。观察3种激发态的空穴–电子分布图(图3)可知,3种激发过程均伴随局部的电荷转移(与硝基相连的O和与中心C原子连接的亚甲基上电子主要向硝基处移动)特征。
表2 基态到9、11、10态的、r、、、coul、HDI和EDI指数数值

Tab.2 Indexes of D, Sr, H, t, Ecoul, HDI and EDI from the ground state to excited states S9, S11, and S10
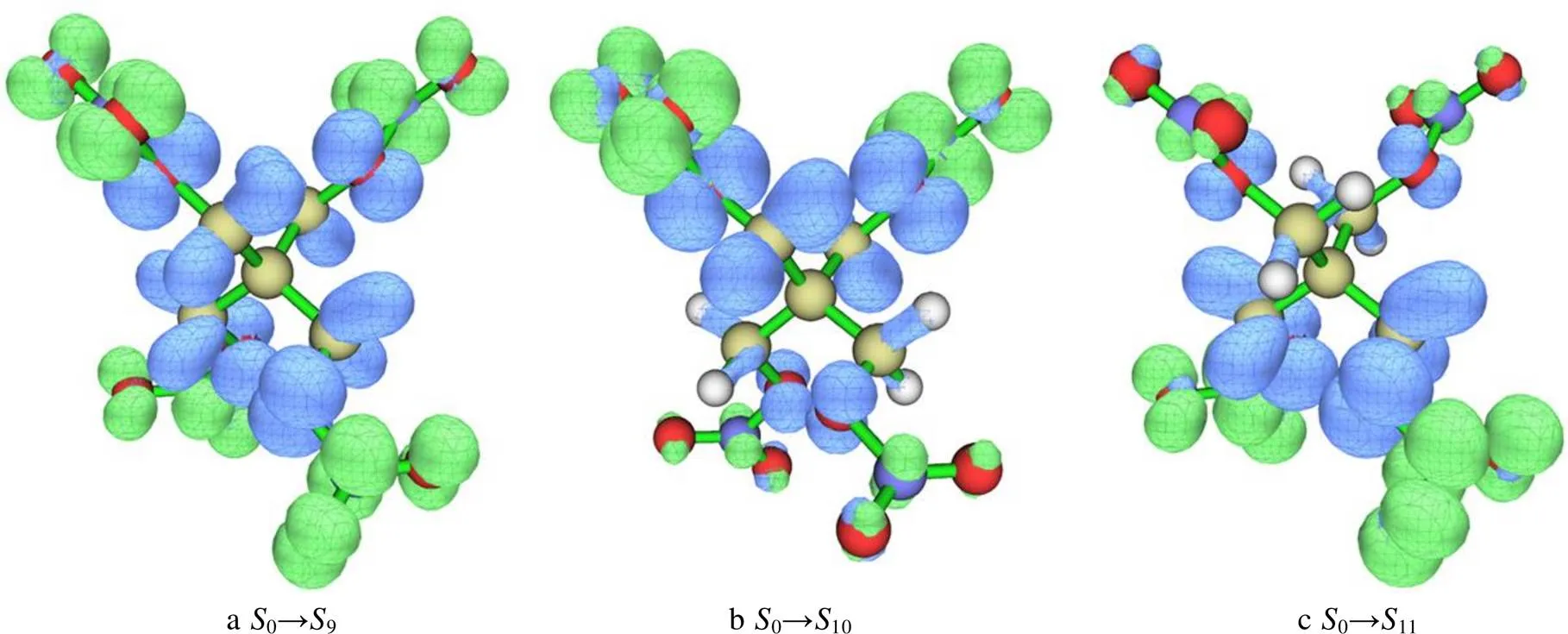
图3 在等值面数值为0.002(a. u.)时基态到S9、S10、S11的空穴(深色)和电子(浅色)分布
从图3可以看出,大部分电子位于PETN的硝基部分,而空穴位于C原子的两侧,0→9态的整体激发特征更为明显,并且10、11态的空穴–电子分布具有明显的对称特征,这与上文所分析的2个激发态为简并态相对应。在指数分析中发现,除EDI参数外,其他参数均相等,同样体现了2个激发态简并。同时,空穴–电子分布图明显可见PETN单分子的3种激发态都具有从中心向四周电荷转移激发的特征,并且空穴集中于与硝基相连的O原子和与中心C原子相连的亚甲基。如图3所示的局部电荷转移可能会引发PETN内O—NO2键强度的变化。
2.2 PETN主要激发过程分析
炸药中最弱键断裂通常被认为是炸药起爆的一个重要特征[22]。一般认为,PETN的引发键为O—NO2键[23-25],研究PETN激发态中O—NO2键的稳定性具有重要意义,同时引发键稳定性的变化也是PETN吸收特定波长并激发至激发态的重要响应体现。2.1节中对PETN主要贡献的3个激发态的电子激发特征进行了分析,其中PETN中局域电荷转移或许会改变O—NO2的强度。在以往的研究中认为,在特征波长的作用下,会降低炸药的起爆能量[8]。因此,本文对PETN分子3种激发态O—NO2键的Mayer键级与Laplace键级进行了计算,计算结果见表3。
PETN基态O—NO2的Mayer键级与Laplace键级分别为0.884和0.285。由表3可知,几种激发态下,PETN分子最弱键的键级均有所降低,即PETN分子在吸收波长为187.00 nm(9)和186.92 nm(10、11)的光后,激发至激发态时,分子内O—NO2键的稳定性被削弱。
表39、10、11激发态下PETN分子O—NO2键的Mayer键级、Laplace键级
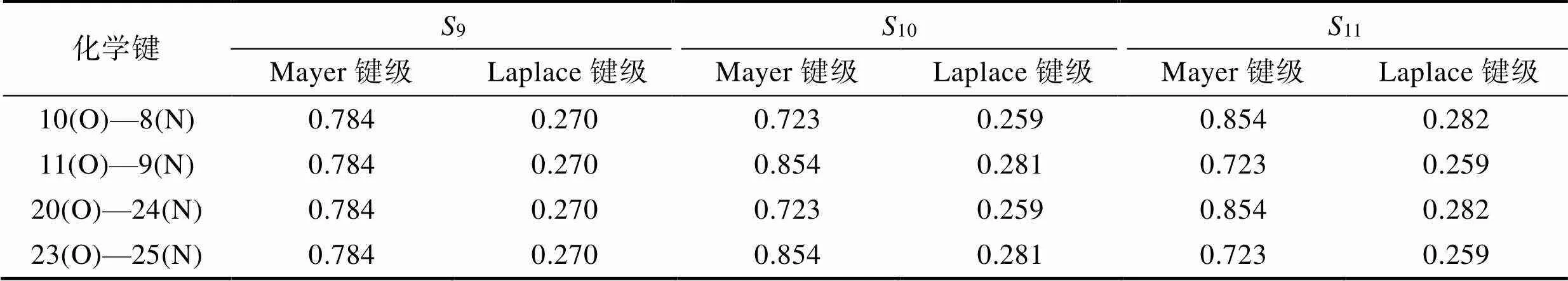
Tab.3 O—NO2 Mayer and Laplace bond orders of PETN molecules in excited states S9, S11, and S10
2.1节的分析中,3种激发态均包含一定程度的整体激发和电荷转移激发特征,3种主要激发过程中,PETN分子内电子的转移可能影响PETN最弱键的稳定性。通过分析O—NO2键级发现,3种主要贡献的激发过程都会降低O—NO2的稳定性。为对此过程进行进一步分析,使用IFCT方法对PETN的激发过程中片段间的电子转移进行了计算。IFCT分析基于空穴与电子贡献来衡量体系中片段间电子的转移量。将PETN分子内的4个硝基和与硝基相连的4个O原子,以及与1号C原子相连的4个亚甲基视为研究对象(原子编号如图1所示),IFCT计算结果见表4。
表4 3种激发过程下PETN分子内片段上电子的变化
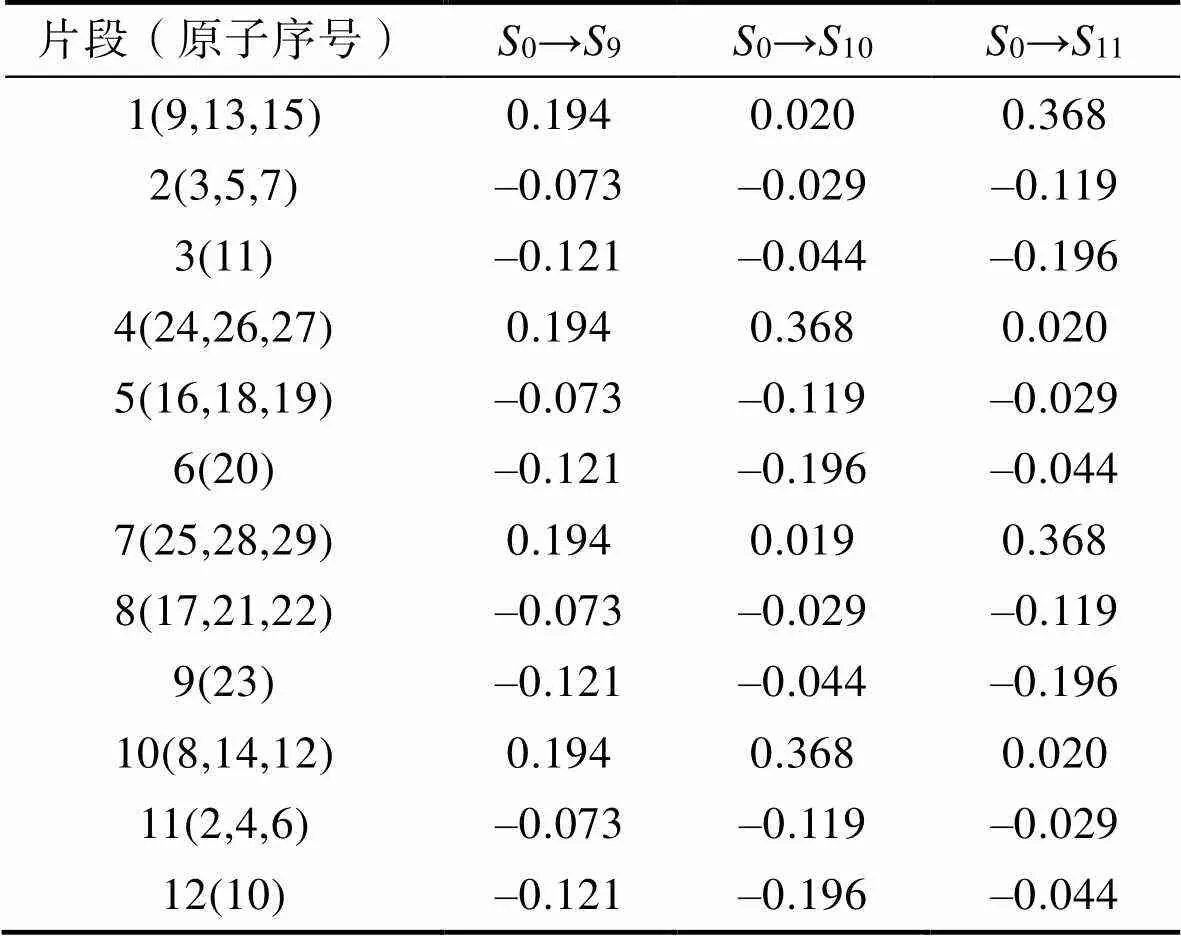
Tab.4 Variation of electrons on fragments in PETN molecules under three excitation processes
表4中片段1、4、7、10表示PETN的硝基,2、5、8、11表示亚甲基,3、6、9、12表示与硝基相连的O原子。在9态中,PETN硝基基团均得到0.194e,与9态明显由中心向四周CT的特征相吻合,除硝基外,PETN其余基团都发生了电子的转移,并且与硝基相连的O原子的电子转移占主导(61.7%)。在10态中,PETN分子4、10号硝基得到0.368e,6、12号氧原子各转移0.196e,5、11号亚甲基各转移0.119e。11态与10态类似,1、7号硝基基团得到0.368e,而3、9号氧原子各转移0.196e,2、8号PETN亚甲基各转移0.119e。PETN其他片段如5、11号亚甲基各转移0.029e,6、12号氧原子各转移0.044e,电荷转移数值较小,对分子内总的电子转移贡献程度较低。3种激发态中,电子主要由与硝基相连的O原子提供,而电子主要转移至硝基上。
以上对PETN各片段间的电子转移分析表明,电子主要来自于PETN分子内与硝基相连的氧原子。进一步使用IFCT的方法对3个激发态中与硝基相连的O原子与硝基之间的电子净转移进行了分析。在9态中,每个与硝基相连的O原子向此硝基净转移的电荷为0.092 1e,这与空穴–电子的分析结果相吻合;在10态中,3、9号O原子分别向4、10号硝基基团净转移的电子均为0.019 6e;在11态中,6、12号O原子分别向1、7号硝基转移0.019 6e,都与空穴–电子分布图分析相吻合。观察图3可知,与硝基相连的O原子上,空穴分布在氧原子两侧,同时硝基上的电子分布具有明显的对称面。结合IFCT分析可知,氧原子与硝基的n→pi*跃迁,促使与硝基相连的O原子发生电子转移,并导致O原子携带的电荷降低,同时这也是PETN内O—NO2键削弱的重要原因。
综上所述,PETN分子内0→9、0→10和0→11这3个激发过程,以n→pi*跃迁特征为主导的电子转移会导致PETN引发键稳定性的降低,即PETN在吸收特定的紫外光并激发至激发态(9、10、11)会促使分子的稳定性降低。
3 结论
本文基于TDDFT理论对PETN在紫外光下的稳定性退化机制进行了研究,首先基于PETN在pbe1pbe/6-311G**水平下的50个激发态计算绘制吸收光谱,并与实验测试数据对比,其中理论计算峰位置小于实验吸收峰位置8.4 nm。
使用空穴–电子方法,对PETN紫外吸收光谱贡献较高的3种激发态的激发特征进行了分析。3种激发态的空穴集中于与硝基相连的O原子和与中心C原子相连的亚甲基,激发过程可能会促使PETN内O—NO2键强度的变化。3种激发态均存在一定程度整体激发和由中心向四周电荷转移激发的特征。
对PETN分子激发态与基态的引发键O—NO2的Mayer与Laplace键级进行了计算,计算结果表明,3种起到主要贡献的激发过程都会降O—NO2键的强度,即PETN在吸收特定波长的紫外光后,激发至激发态,这一过程会削弱引发键的稳定性。
使用IFCT方法对PETN分子片段间的电子转进行了分析,分析结果表明,与硝基相连的氧原子和硝基之间的n→pi*跃迁是PETN引发键稳定性削弱的重要原因。
[1] 董海山, 周芬芬. 高能炸药及相关物性能[M]. 北京: 科学出版社, 1989.
DONG Hai-shan, ZHOU Fen-fen. Performance of High Explosives and Correlates[M]. Beijing: Science Press, 1989.
[2] 王宇锐, 范新中, 王晓鹏, 等. 激光火工品系统在战术导弹领域的应用前景与展望[J]. 导弹与航天运载技术, 2021(6): 15-20.
WANG Yu-rui, FAN Xin-zhong, WANG Xiao-peng, et al. Application Prospect of Laser Ignition System in Tactical Missile Field[J]. Missiles and Space Vehicles, 2021(6): 15-20.
[3] YANG L C, MENICHELLI V J. Optically Detonated Explosive Device: US3812783[P]. 1974-05-28.
[4] 王宇, 魏超, 张嵩. 钝感火工品中新技术、新含能材料研究进展[J]. 科技导报, 2013, 31(10): 74-79.
WANG Yu, WEI Chao, ZHANG Song. Developing Status of New Techniques and New Energetic Materials in Insensitive Pyrotechnics[J]. Science & Technology Review, 2013, 31(10): 74-79.
[5] 梅崴, 徐宇新, 邢朝洋, 等. 激光点火系统用1×2 MEMS光开关研究[J]. 导航与控制, 2017, 16(1): 85-90.
MEI Wei, XU Yu-xin, XING Chao-yang, et al. Research of 1×2 MEMS Optical Switch for Laser Ignition System[J]. Navigation and Control, 2017, 16(1): 85-90.
[6] TARZHANOV V I, ZINCHENKO A D, SDOBNOV V I, et al. Laser Initiation of PETN[J]. Combustion, Explosion and Shock Waves, 1996, 32(4): 454-459.
[7] 盛涤伦, 朱雅红, 陈利魁, 等. 激光与含能化合物相互作用机理研究[J]. 含能材料, 2008, 16(5): 481-486.
SHENG Di-lun, ZHU Ya-hong, CHEN Li-kui, et al. Interactional Mechanism between Laser and Energetic Compound[J]. Chinese Journal of Energetic Materials, 2008, 16(5): 481-486.
[8] ALUKER E D, KRECHETOV A G, MITROFANOV A Y, et al. Laser Initiation of Energetic Materials: Selective Photoinitiation Regime in Pentaerythritol Tetranitrate[J]. The Journal of Physical Chemistry C, 2011, 115(14): 6893-6901.
[9] 刘建, 熊鹰, 蒋小华, 等. 含能材料激光诱导起爆特征波长解析[J]. 激光技术, 2013, 37(6): 816-819.
LIU Jian, XIONG Ying, JIANG Xiao-hua, et al. Characteristic Wavelength Analysis for Laser-Induced Initiation in Energetic Material[J]. Laser Technology, 2013, 37(6): 816-819.
[10] TSYSHEVSKY R V, SHARIA O, KUKLJA M M. Energies of Electronic Transitions of Pentaerythritol Tetranitrate Molecules and Crystals[J]. The Journal of Physical Chemistry C, 2014, 118(18): 9324-9335.
[11] TARZHANOV V I, SDOBNOV V I, ZINCHENKO A D, et al. Laser Initiation of Mixtures of PETN and Aluminum by a Deposit[J]. Combustion, Explosion, and Shock Waves, 2017, 53(6): 724-729.
[12] ADUEV B P, NURMUKHAMETOV D R, NELYUBINA N V, et al. Initiation Thresholds and Dynamic Characteristics of an Explosion for Thin Samples of PETN-Al Composites under Laser Irradiation[J]. Technical Physics, 2019, 64(6): 858-864.
[13] ADUEV B P, NURMUKHAMETOV D R, ZVEKOV A A, et al. Laser Initiation of PETN-Based Composites with Additives of Ultrafine Aluminium Particles[J]. Combustion, Explosion, and Shock Waves, 2016, 52(6): 713-718.
[14] 湛赞, 谭明, 严楠, 等. 半导体激光起爆炸药试验与仿真研究[J]. 弹箭与制导学报, 2020, 40(5): 10-14.
ZHAN Zan, TAN Ming, YAN Nan, et al. Experimental and Simulation Study of Laser Diode Detonation[J]. Journal of Projectiles, Rockets, Missiles and Guidance, 2020, 40(5): 10-14.
[15] CADY H H, LARSON A C. Pentaerythritol Tetranitrate II: Its Crystal Structure and Transformation to PETN I; An Algorithm for Refinement of Crystal Structures with Poor Data[J]. Acta Crystallographica Section B Structural Crystallography and Crystal Chemistry, 1975, 31(7): 1864-1869.
[16] FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. 2009. Gaussian 09[R]. Inc. USA: Wallingford CT.
[17] LU Tian, CHEN Fei-wu. Multiwfn: A Multifunctional Wavefunction Analyzer[J]. Journal of Computational Chemistry, 2012, 33(5): 580-592.
[18] LIU Ze-yu, LU Tian, CHEN Qin-xue. An Sp-Hybridized All-Carboatomic Ring, Cyclo[18]Carbon: Electronic Structure, Electronic Spectrum, and Optical Nonlinearity[J]. Carbon, 2020, 165: 461-467.
[19] YANG Jun-qing, ZHANG Gu-dan, ZHANG Jian-guo, et al. New Perspectives on the Laser Initiation for Metal Tetrazine Complexes: A Theoretical Study[J]. Physical Chemistry Chemical Physics: PCCP, 2021, 24(1): 305-312.
[20] LIU Run-ze, SHANG Fang-jian, XIONG Ying, et al. A Combined Experimental and Theoretical Investigation of the Excited-State Dynamics of 2, 4, 6-Trinitrotoluene (TNT) in DMSO Solvent[J]. Physical Chemistry Chemical Physics: PCCP, 2021, 23(36): 20718-20723.
[21] HILBORN R C. Einstein Coefficients, Cross Sections,Values, Dipole Moments, and all that[J]. American Journal of Physics, 1982, 50(11): 982-986.
[22] 曹霞, 向斌, 张朝阳. 炸药分子和晶体结构与其感度的关系[J]. 含能材料, 2012, 20(5): 643-649.
CAO Xia, XIANG Bin, ZHANG Chao-yang. Review on Relationships between the Molecular and Crystal Structure of Explosives and Their Sensitivities[J]. Chinese Journal of Energetic Materials, 2012, 20(5): 643-649.
[23] LANDERVILLE A C, OLEYNIK I I, WHITE C T, et al. Reactive Molecular Dynamics of Detonating Petn[J]. AIP Conference Proceedings, 2009, 1195(1): 813-816.
[24] WU C, REE F, YOO C S. A Quantum Mechanical Molecular Dynamics Study of Binary Collisions of Pentaerythritol Tetranitrate (PETN): Its Correlation to Shock Sensitivity[J]. Propellants, Explosives, Pyrotechnics, 2004, 29(5): 296-303.
[25] RICE B M, SAHU S, OWENS F J, et al. Density Functional Calculations of Bond Dissociation Energies for NO2Scission in some Nitroaromatic Molecules[J]. Journal of Molecular Structure: Theochem, 2002, 583(1/2/3): 69-72.
Qualitative Effects of Ultraviolet Light on PETN Based on TDDFT
ZHANG Bao-sen1, ZHANG Shu-hai1, GOU Rui-jun1, CHEN Ya-hong1, ZHU Shuang-fei1, MA Kun2
(1. School of Environmental and Safety Engineering, North University of China, Taiyuan, 030051, China; 2. Shaanxi Applied Physics-chemistry Research Institute, Xi'an 710061, China)
The work aims to study the stability degradation mechanism of PETN explosives under the action of ultraviolet light. Based on the TDDFT (time-dependent density functional) theory, 50 excited states of PETN molecules at the pbe1pbe/6-311G** level were calculated and the ultraviolet absorption spectrum was drawn according to the calculation results, and the excitation characteristics of the three excited states of the maximum absorption peak (the total contribution rate of the three excited states of S9, S10, and S11to the maximum absorption peak was 97.31%) were analyzed by the hole-electron method. These three excited states were set as the research objects to study the Mayer and Laplace bond orders of PETN molecules' weak bond after absorbing specific ultraviolet light. Finally, the electron transfer of PETN molecules excited to the excited states were described based on the IFCT (Interfragment Charge Transfer) method. The maximum absorption peak position of the PETN ultraviolet absorption spectrum calculated in this work was 186.6 nm, which was less than 8.4 nm of the absorption peak measured in experiment. Among the three excited states that contributed the most to this absorption peak, the S9state had the highest contribution of 48.27%, and the other two excited states S10and S11were degenerate states with the same contribution of 24.52%. According to the results of the hole-electron analysis, all the three excited states had the characteristics of overall excitation and partial charge transfer. When the PETN molecules absorbed ultraviolet light of a specific wavelength (187.00 nm and 186.92 nm) and then was excited to the corresponding excited state, the Mayer and Laplace bond orders at O—NO2bonds decreased. The IFCT analysis shows that the change of Mayer bond order is guided by the change from n→Pi* on O—NO2. This effect may reduce the stability of PETN molecules.
PETN; ultraviolet; TDDFT; absorption spectrum; molecular stability; excited state
TJ450
A
1672-9242(2022)12-0048-06
10.7643/ issn.1672-9242.2022.12.008
2022–08–16;
2022–11–21
2022-08-16;
2022-11-21
陕西应用物理化学研究所应用物理化学重点实验室基金(6142602200304)
Science and Technology on Applied Physicial Chemistry Laboratory, Shaanxi Applied Physics-Chemistry Research Institute (6142602200304)
张宝森(1998—),男,硕士研究生,主要研究方向为含能材料相关计算化学。
ZHANG Bao-sen (1998-),Male, Postgraduate, Research focus: related computational chemistry of energetic materials.
张树海(1969—),男,博士,教授,主要研究方向为新型火工药剂技术及工程爆破。
ZHANG Shu-hai (1969-), Male, Doctor, Professor, Research focus: new initiating explosive technology and engineering blasting.
张宝森,张树海,苟瑞君, 等. 基于含时密度泛函理论的紫外光对太安的定性影响[J]. 装备环境工程, 2022, 19(12): 048-053.
ZHANG Bao-sen, ZHANG Shu-hai, GOU Rui-jun, et al. Qualitative Effects of Ultraviolet Light on PETN Based on TDDFT[J]. Equipment Environmental Engineering, 2022, 19(12): 048-053.
责任编辑:刘世忠
