1q21.1微缺失/微重复胎儿的临床表型和遗传学分析
2023-01-10傅婉玉庄建龙王元白江矞颖肖珊珊李燕青
傅婉玉,庄建龙,王元白,江矞颖,肖珊珊,李燕青
(泉州市妇幼保健院·儿童医院产前诊断中心,泉州 362000)
基因组拷贝数变异(copy number variants,CNVs)是遗传上常见的染色体畸变类型,与某些疾病发生紧密相关[1]。在一些复杂性遗传疾病中,CNVs决定了不同个体对于相同外界刺激的差异反应以及对于相同疾病的差异易感性。研究显示,1q21.1微缺失/微重复是发生在1号染色体长臂上的CNVs[2],1q21.1微缺失/微重复的个体具有广泛的临床表现,包括头面部异常、发育迟缓、智力残疾、行为异常(包括自闭症和注意缺陷多动障碍等)、先天性心脏畸形和其他畸形特征[3-5],但也有一些1q21.1微缺失/微重复的携带个体并无明显的临床症状,目前相关机制尚不清楚[5]。1q21.1微缺失/微重复导致的体征和症状可能与该区域几个基因的CNVs有关。然而,目前仍缺乏对于1q21.1发生微缺失/微重复区段的基因CNVs与临床症状之间关系的系统研究,且1q21.1微缺失/微重复产前诊断的病例报道有限。本研究纳入8例1q21.1拷贝数异常的胎儿,分析其产前超声特征和遗传病表型,探讨1q21.1微缺失/微重复在胎儿期的临床表型。
1 资料与方法
1.1 研究对象 回顾分析于泉州市妇幼保健行染色体核型分析及单核苷酸多态性微阵列(single nucleotide polymorphism array,SNP-array)检测的产前诊断病例3748例,其中8例产前诊断确认为1q21.1微缺失/微重复胎儿。孕妇行介入性产前诊断和SNP-array检测前均充分知情并签署知情同意书。8例中病例1~7行羊膜腔穿刺术取羊水样本(各30mL),病例8行经腹脐静脉穿刺术取脐血样本(各2mL)。8例孕妇均有产前诊断指征(血清学筛查高风险、胎儿超声结构异常、胎儿超声软指标异常、孕妇高龄等)。
1.2 染色体核型分析 将10mL羊水和1mL脐血进行体外细胞培养,待细胞增殖后收获适量培养细胞进行染色体制备并进行核型分析,各样本均分析了5个核型。染色体的命名依据人类细胞遗传学国际命名体制(ISCN 2020)的标准。
1.3 SNP-array检测 取10mL羊水/1mL脐血样本及夫妻双方外周血各3mL送至第三方检测公司(北京贝康医学检验所)检测,离心收集沉淀,按Affymetrix Cytoscan 750K基因芯片试剂盒(赛默飞世尔,美国)说明书进行操作,提取羊水细胞基因组DNA,经稀释消化、扩增、纯化,将芯片上的探针与用生物素进行标记后的相应待测片段进行杂交、洗涤、结合染色,放入GeneChip®扫描仪(Affymetrix,美国)扫描,采用Chromosome Analysis Suite(ChAS) v4.0软件对扫描检测的荧光信号进行分析。根据美国医学遗传学会指南,通过查询DGV、OMIM、Pubmed、DECIPHER以及UCSC数据库对CNVs的临床意义进行分析。
2 结 果
2.1 染色体核型分析结果 羊水染色体核型分析结果显示,病例1胎儿染色体核型为46,XN,inv(9)(p11;q13),7例胎儿染色体核型均正常。
2.2 SNP-array检测结果 检出1q21.1微缺失/微重复各4例。微缺失分别为病例2~5,片段大小分别为503.5Kb、1.9Mb、1.9Mb和2.0Mb;微重复分别为病例1和病例6~8,片段大小为393.8Kb、1.3Mb、2.1Mb和3.8Mb。8例1q21.1微缺失/微重复区段涉及的全部蛋白编码基因共19个,其中包括已报道的致病基因PRKAB2(*602741),CHD1L(*613039),GJA5(*121013)和GJA8(*600897)(图1)。病例2~3和病例5进行了父母溯源家系验证,其中病例2~3为父源遗传,病例5为新发变异,其余病例未进行溯源验证(表1)。
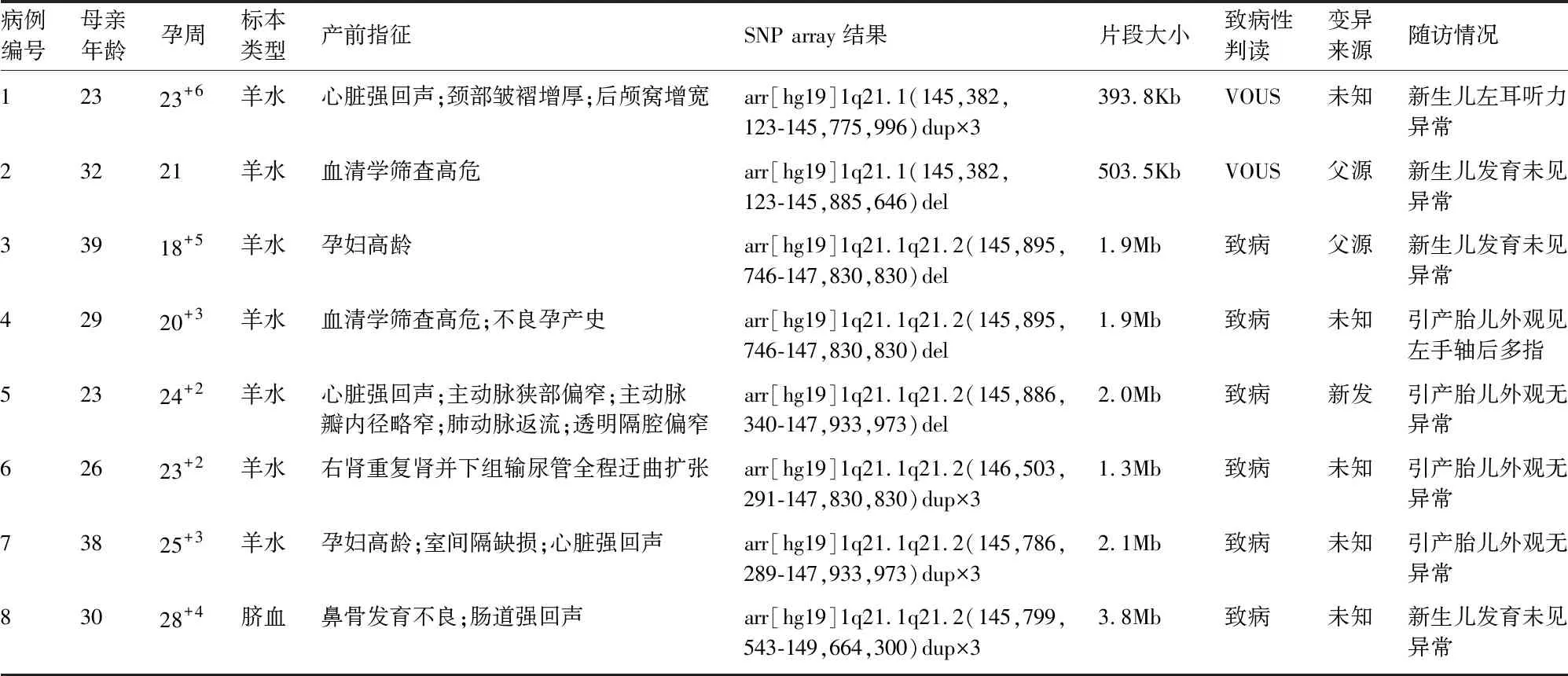
表1 8例1q21.1微缺失/微重复胎儿的临床数据
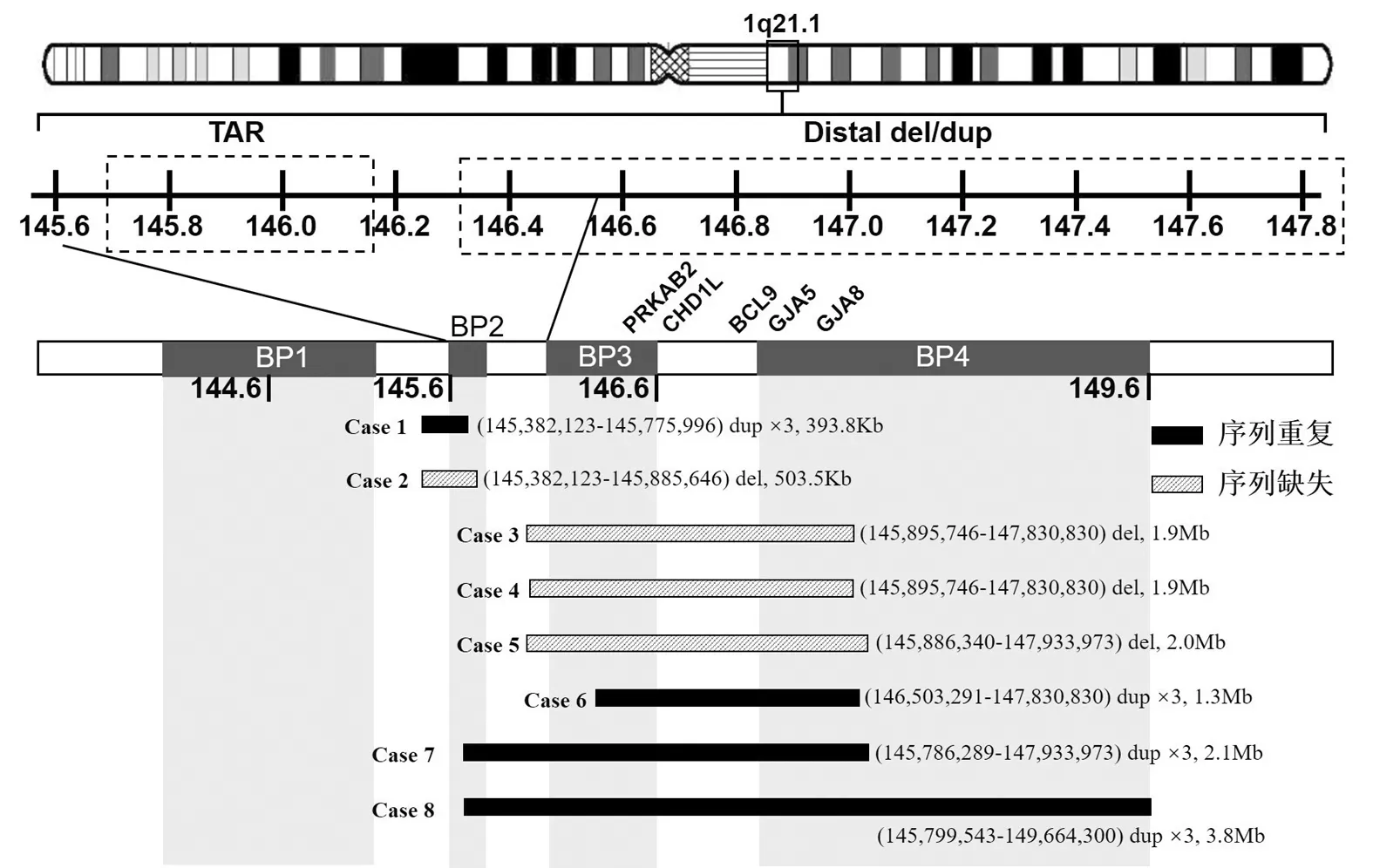
图1 8例1q21.1微缺失/微重复位置
2.3 各病例的其他情况
2.3.1 微重复病例 (1)病例1,孕23+周,超声显示胎儿心脏强回声,颈部皱褶增厚,后颅窝增宽。经充分遗传咨询后选择继续妊娠,于2018年9月15日剖宫产一男婴,出生体重4.5kg,新生儿筛查提示左耳听力异常,2019年1月24日行脑干诱发电位检测左耳听力仍未通过,后续失访。(2)病例6,孕23周,超声显示胎儿右肾重复肾并下组输尿管全程迂曲扩张,经充分遗传咨询后选择终止妊娠,引产胎儿外观无异常。(3)病例7,孕25周,超声显示胎儿心脏室间隔缺损0.38cm,左心室点状强回声,经充分遗传咨询后选择终止妊娠,引产胎儿外观无异常。(4)病例8,孕28周,超声显示胎儿鼻骨发育不良、部分肠管强回声,经充分遗传咨询后选择继续妊娠,于2021年12月8日剖宫产一女婴,目前出生后4+月会翻身,体格发育正常。
2.3.2 微缺失病例 (1)病例2,孕21周,血清学筛查高风险,产前超声未提示明显异常,经充分遗传咨询后选择继续妊娠,于2019年1月足月经阴分娩一女,目前生后3+岁,体格、语言、智力、运动发育均正常。(2)病例3,孕18周,高龄孕妇,产前超声未提示明显异常,经充分遗传咨询后选择继续妊娠,于2021年3月31日足月经阴分娩一男,9个月会喊妈妈爸爸,目前1岁会爬,可扶走,身高76cm,体重8.78kg。(3)病例4,孕20周,血清学筛查高风险,有不良孕产史(既往于2016年经阴分娩一子,患有先天性心脏病:室间隔缺损、房间隔缺损、卵圆孔未闭、肺动脉流速增快、左上腔静脉残存,患儿生后1+岁行心脏手术,术后恢复良好,未行染色体检查)。本次产前超声未提示明显异常,经充分遗传咨询后选择终止妊娠,引产胎儿外观左手轴后多指。(4)病例5,孕24周,超声显示胎儿心脏强回声、主动脉峡部偏窄、主动脉瓣内径偏窄、肺动脉返流、透明隔腔偏窄。经充分遗传咨询后选择终止妊娠,引产胎儿外观无异常。
3 讨 论
在微重复病例和微缺失病例中,患儿产前指标和产后临床症状并无明显相关性,表明1q21.1区段的重复或缺失并不能作为某种临床症状的直接证据。本研究初步发现,重复/缺失片段大小与临床表现之间不能建立联系,即不能依据变异片段大小推测临床表现。本结果进一步佐证了1q21.1微重复/微缺失变异的多变性和致病机制的复杂性[6]。本研究中,4例1q21.1微重复病例和4例1q21.1微缺失病例,各有2例成功妊娠分娩,其余由家属自行选择终止妊娠。
1q21.1是一个复杂的区域,具有多个低拷贝重复系列,这些低拷贝重复序列聚集在一起,形成4个片段重复区域,大小范围为270kb~2.2Mb,使该亚染色体区域成为非等位基因同源重组(non-allelic homologous recombination,NAHR)的热点,从而使该区域易于反复缺失和反复重复。1q21.1微缺失/微重复的染色体断点(BPs)被专门定位到这4个片段重复区域,从着丝粒到端粒的定位分别定为BP1-BP4(图1)[3-4]。发生在近端BP2和BP3之间的微缺失被认为是TAR(Thrombocytopenia-absent Radius)综合征的诱发因素[7-8]。TAR综合征的特征是双侧前臂桡骨缺失和血小板减少。除了双侧桡骨缺失外,尺骨发育不全、肱骨和肩带缺失,其他骨骼异常(包括肋骨和椎体受累)、心脏和泌尿生殖系统畸形也可能出现[9]。本文中2例涉及TAR区域的近端微缺失/微重复(分别各1例),病例1超声显示心脏强回声、颈部皱褶增厚、后颅窝增宽,没有表现出桡骨不全和尺骨发育异常等骨骼发育障碍;病例2超声正常,新生儿没有任何发育异常的表型。2例病例均未体现TAR综合征的典型症状,且2例病例临床表现也有差异,这种差异现象可能与1q21.1微重复/微缺失表型的异质性或不全外显性有关。值得注意的是,病例1出生后伴有先天性听力障碍,但由于本文只关注了1q21.1区段的微重复/微缺失,因此不排除其他基因突变或环境因素导致先天性听力障碍的可能性。
涉及远端BP4的1q21.1微缺失/微重复包含两类,Ⅰ类是只包含远端BP3-BP4的1q21.1区域,约1.35Mb大小,Ⅱ类为发生在BP2-BP4的1q21.1区域,范围较大,从3.0Mb~3.9Mb[3]。远端1q21.1微缺失/微重复通常表现为轻微发育迟缓、颅面异常特征、精神行为异常和先天性发育异常[可能体现为先天性心脏病、眼睛异常(小眼症、视网膜脉络膜和虹膜缺损、斜视、各种类型的白内障)、骨骼和泌尿生殖系统畸形];少数患者中表现出精神行为异常,如自闭症谱系障碍,精神分裂症和注意力缺陷多动障碍、在某些情况下,癫痫也有发作(15%)[3-4]。但这些表型中精神行为异常、发育迟缓、癫痫等表型具有年龄依赖性,无法在宫内诊断。本文中病例3~8均为远端微缺失/微重复,其中病例3~5的微缺失和病例7~8的微重复发生在BP2-BP4之间,为Ⅱ类远端微缺失/微重复。根据产前超声结果看,病例7的胎儿心脏发育异常,病例8鼻骨发育不良和肠道回声增强,2例患者的临床表现都有典型的1q21.1微缺失/微重复的症状,但病例8的新生儿未见异常,可能与该疾病的不全外显性有关[10]。病例3无明显异常,其变异来源的父亲也没有明显的临床表型,也可能是因为不全外显性。病例4引产胎儿外观左手轴后多指。文献中曾报道1例涉及远端1q21.1微缺失胎儿出生后表现左足轴后多指畸形[11],遗传自表型正常的父亲,该表型是否与1q21.1微缺失有关还有待更多的病例被识别以及进一步的深入研究。病例6为只发生在远端(BP3-BP4)的微重复/微缺失,为I类远端微重复,肾脏发育异常,属于泌尿系统异常的典型案例。
远端1q21.1区域内存在许多关键基因,主要包括剂量敏感的PRKAB2、CHD1L、BCL9、GJA5和GJA8。PRKAB2和CHD1L基因的编码产物分别参与AMP(一磷酸腺苷)和ATP(三磷酸腺苷)的活动过程,PRKAB2和CHD1L的单基因功能障碍导致患者在早期发育过程中更易发生发育障碍[12]。PRKAB2和BCL9(Wnt信号中的一个功能基因[13])是精神分裂症、自闭症、孤独症及注意缺陷多动障碍等精神疾病的易感基因[14],这可能也是部分1q21.1微重复/微缺失易患有精神疾病临床表现的主要原因,但该类精神疾病的发生受到年龄和环境因素的影响,因此这两种基因可作为精神分裂症的候选基因以评估发生精神疾病的风险。病例3和病例8分别为涉及PRKAB2和BCL9缺失和重复的变异的存活案例,有必要继续随访以充实临床数据。本研究中病例3~8均涉及CHD1L基因缺失/重复变异,仅有病例6表现出肾脏发育障碍(右肾重复肾并下组输尿管全程迂曲扩张),这与文献报道[15]有一定的共同点,即涉及CHD1L基因变异的1q21.1微重复/微缺失可能导致肾脏发育障碍,如双肾发育不良、单侧肾缺如伴巨输尿管,且症状表现出家族遗传性。但病例3~5和病例7~8并未发现肾脏发育障碍,表明CHD1L基因变异导致的肾脏发育障碍的临床表型存在不完全显性或可变表现度。此外,CHD1L基因杂合错义变异的患者表现为先天性肾脏和泌尿系异常(Congenital anomalies of the kidney and urinary tract,CAKUT)[16-17],提示CHD1L可作为一个新的先天性肾脏发育障碍的候选基因。GJA5和GJA8基因属于连接蛋白基因家族的成员,分别在心脏发育和眼睛晶状体发育过程中发挥细胞间隙连接作用[18]。从功能方面讲,1q21.1微重复/微缺失患者发生心脏发育障碍的原因可能与GJA5基因发生CNVs有关[6,18-20]。本研究中病例5、7,超声均提示胎儿不同程度的心脏发育高危,可能与GJA5基因的CNVs有关,但病例3、4、6、8没有提示心脏异常,进一步证实了该区段的微重复/微缺失作用机制的复杂性。GJA8的CNVs与患者发生眼部发育障碍、白内障或眼动障碍之间也不存在直接关系,如病例8,尽管GJA8基因拷贝数变异,但新生儿出生时未发生先天性眼疾或眼部功能障碍,有待进一步随访。
综上,尽管上述5种基因被认为是1q21.1区段的重点基因,也作为变异致病性的重要参考,但由于1q21.1本身结构的复杂性和变异的多变形和复杂性,加上涉及基因不全外显和可变表现度,这五种单基因CNVs作为致病性重要参考具有不全面性。除了上述的5种重点基因,1q21.1远端还包括其他十余种蛋白编码基因,这些基因的变异也可能是导致患者发育异常的关键因素。研究人员也正在积极开展相关研究,如挖掘1q21.1区域CNVs正相关的候选基因(GWASs)以及表型严重程度、基因组外显性和遗传方面的变化[5]。一项调查结果显示,在小头畸形的案例中,女性比男性更易患小头畸形症[21],但仍未能建立明确的变异与致病之间的关系。因此,在继续筛找候选基因并系统研究这些基因的CNVs功能机制领域仍存在大量空白。
结合目前有关1q21.1微缺失/微重复胎儿的报道,患者在产前可出现各种不同生理系统超声异常,较常见的宫内表型为泌尿系畸形和心脏畸形[22-23],其他包括神经管畸形[24]、小头畸形[21]、多指/趾[11]、鼻骨发育不良[25]、NT增厚[26-27]等。产前超声显示胎儿存在较明确结构畸形时,更倾向于终止妊娠。不完全外显率和表达度差异为产前诊断的咨询带来了挑战,特别是对于产前无法确定的神经发育表型(主要指自闭症等精神疾病),胎儿在宫内未出现明显异常时往往不能依靠产前超声进行诊断。因此,产前筛查和检测中,纳入SNP-array检测可有效检出1q21.1微缺失和微重复,有助于提高1q21.1微重复/微缺失患者的检出率,以及建立变异机制与临床表型的对应关系,对于早期发现、认识和判断其致病性也具有重要的参考意义,同时能为患者的妊娠结局、预后及治疗提供重要的依据。
