gRNA二级结构对拟南芥HD2基因CRISPR/Cas9编辑率的影响
2023-01-09黄轩雅欧艺鹏魏淑怡
黄轩雅,欧艺鹏,魏淑怡,林 娟
(1.复旦大学 生命科学学院,上海 200438;2.复旦大学 遗传工程国家重点实验室,上海 200438)
突变体的获得途径是研究基因功能的重要基础。获得突变体的方法有物理方法[1]、化学方法[2]和基因工程方法[3]。前两种方法对基因的改变具有随机性,不能够对靶基因进行直接修饰,因此其应用受到了很大的限制。基因工程方法是近年来发展的一种新型的突变体获得技术,尤其是基因组编辑技术已成为基因改造和研究的主要手段,并已成功在多种植物中得到普遍应用[4]。CRISPR/Cas9(Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-Associated Proteins 9)是继锌指核酸酶技术(Zinc-Finger Nucleases, ZFNs)[5]和转录激活因子样效应物核酸酶技术(Transcription Activator-Like(TAL) Effector Nucleases, TALENs)[6]之后发展的第3代基因组定点编辑技术,是目前最新和最高效的基因编辑技术。这一技术首先在人类和小鼠细胞中成功对部分基因实现了编辑[7],之后在模式植物拟南芥[8]、烟草[9]、水稻[10]和一些经济植物[11-12]中获得了广泛的应用,成功实现了靶点的突变。
CRISPR是指一段具有成簇的规律间隔的短回文重复序列,通过CRISPR系统将目标序列切断后,生物个体会启动自身的修复机制,包括同源重组修复和非同源重组修复,在修复后产生插入、缺失等突变,导致目标基因沉默。其作用原理是核酸内切酶Cas9蛋白在gRNA(single-guide RNA)引导下,在目标基因的特定区域对特定的DNA进行定点切割,造成DNA双链的断裂,细胞实行自主修复机制,从而实现特定位点的定向编辑。虽然CRISPR/Cas9技术在多种植物的基因编辑中取得了成功,然而这一编辑系统的编辑率却因物种不同、基因不同而存在明显差异,因此如何提高靶基因的编辑率是该技术的关键点,也是研究者们重点关注的焦点。提高CRISPR/Cas9技术基因编辑率主要考虑两个方面,一是gRNA的设计;二是Cas蛋白的改造。CRISPR/Cas9系统工作的核心在于人工设计gRNA,gRNA是由crRNA(crispr RNA)和反式激活RNA即tracrRNA(trans-activating CRISPR RNA)两种非编码RNA通过碱基配对结合而成[13],目前已经将其融合为一条链,仍称为gRNA,因此人工设计的gRNA是影响CRISPR/Cas9技术基因编辑率的主要因素,这主要是对靶位点序列的设计。虽然研究者们设计了多款专门用于gRNA设计的在线软件[14-16],这些软件能够筛选出评分较高的gRNA,为研究者们设计gRNA提供了一条途径。但仅仅根据设计软件,筛选使用评分最高的gRNA来构建载体,后续的基因编辑率仍然得不到有效的保障[17]。目前研究发现gRNA序列上优化的GC(Guanine-Cytosine)含量对基因的编辑率会产生一定的影响,高GC含量可使gRNA与基因组DNA的杂交趋于稳定[18],而低GC含量可降低脱靶效应[19]。因此研究gRNA的结构与基因编辑的关系对于提高编辑率具有重要的意义。
本研究从gRNA的二级结构入手,选用的载体为2015年报道的CRISPR载体,该载体的策略是通过具有串联排列的tRNA-gRNA结构的合成基因,利用植物体内的RNase P和RNase Z识别出tRNA-gRNA,并将其切割加工成5’靶向序列的gRNA,从而释放gRNA,再由gRNA去引导Cas9蛋白去编辑目的基因[20]。由于tRNA-gRNA后加入的靶位点序列不同,会形成不同的二级结构,也会影响基因的编辑率。本研究选择的靶标基因为植物组蛋白去乙酰化酶(Histone Deacetylase, HDAC)基因家族中一个植物特有的HD2(HD-2 type protein)亚家族基因,通过设计这一亚家族中3个基因不同的CRISPR/Cas9靶点,观察不同的RNA(gRNA+靶点序列)的二级结构对基因编辑率的影响。HD2亚家族在拟南芥中共包含4个基因,分别称为HD2A,HD2B,HD2C和HD2D[21]。之前研究显示HD2基因的过表达或突变体植株具有容易观察的表型,如: 拟南芥HD2A过表达的转基因植株具有多种发育异常,包括叶片卷曲,开花延迟和种子发育中断等[22]。HD2B的CRISPR基因编辑植株的叶片也表现为窄而尖的叶子[23]。HD2D基因的突变体会导致叶片少而小,HD2D过表达的转基因植株叶片多且大[24]。从拟南芥的叶片的变化可以直接看出HD2基因是否被编辑。因此选择HD2基因作为验证不同tRNA-gRNA二级结构影响CRISPR/Cas9编辑率的靶基因,可为CRISPR/Cas9技术的广泛应用提供一定的基础。
1 材料和方法
1.1 植物材料
拟南芥野生型植株Col-0(Columbia生态型),突变体植株hd2b-1(SALK_049380C)和hd2d-1(SALK_104071C)均购自Arabidopsis Biological Resource Center at Ohio State University(ABRC, http:∥abrc.osu.edu),纯合的突变体植株根据SIGnAL(Salk Institute Genomic Analysis Laboratory, http:∥signal.salk.edu/tdnaprimers.2.html)提供的方法及引物序列进行检测。所有植物材料生长条件一致,均培养在恒温温室(22 ℃),湿度65%,光周期为16 h光照,8 h黑暗。
CRISPR/Cas9编辑植株:hd2b-2(HD2B靶点A的G碱基插入),hd2d-2(HD2D靶点A的A碱基缺失)。
1.2 菌株及质粒
大肠杆菌EscherichiacoliDH5α菌株和根瘤农杆菌AgrobacteriumtumefaciensGV3101菌株购自唯地生物公司。PTG模板质粒: pLFC294(pGTR-sp)和CRISPR载体质粒: Pro: 35S: SpCas9来自河南大学陆平利教授的馈赠。
1.3 方法
1.3.1 CRISPR载体构建
根据HD2A,HD2B,HD2D基因的序列,使用网站https:∥www.genome.arizona.edu/crispr/CRISPRsearch.html,对每个基因分别设计两个不同的靶点,分别称为靶点A(Target A)和靶点B(Target B),具体靶点位置如图1(a)(见第148页)所示。通过MEGA软件分别对3个基因的靶点A和靶点B的序列进行了比对,发现不同基因靶点位置的序列有一定的差异(图1(b),见第148页)。
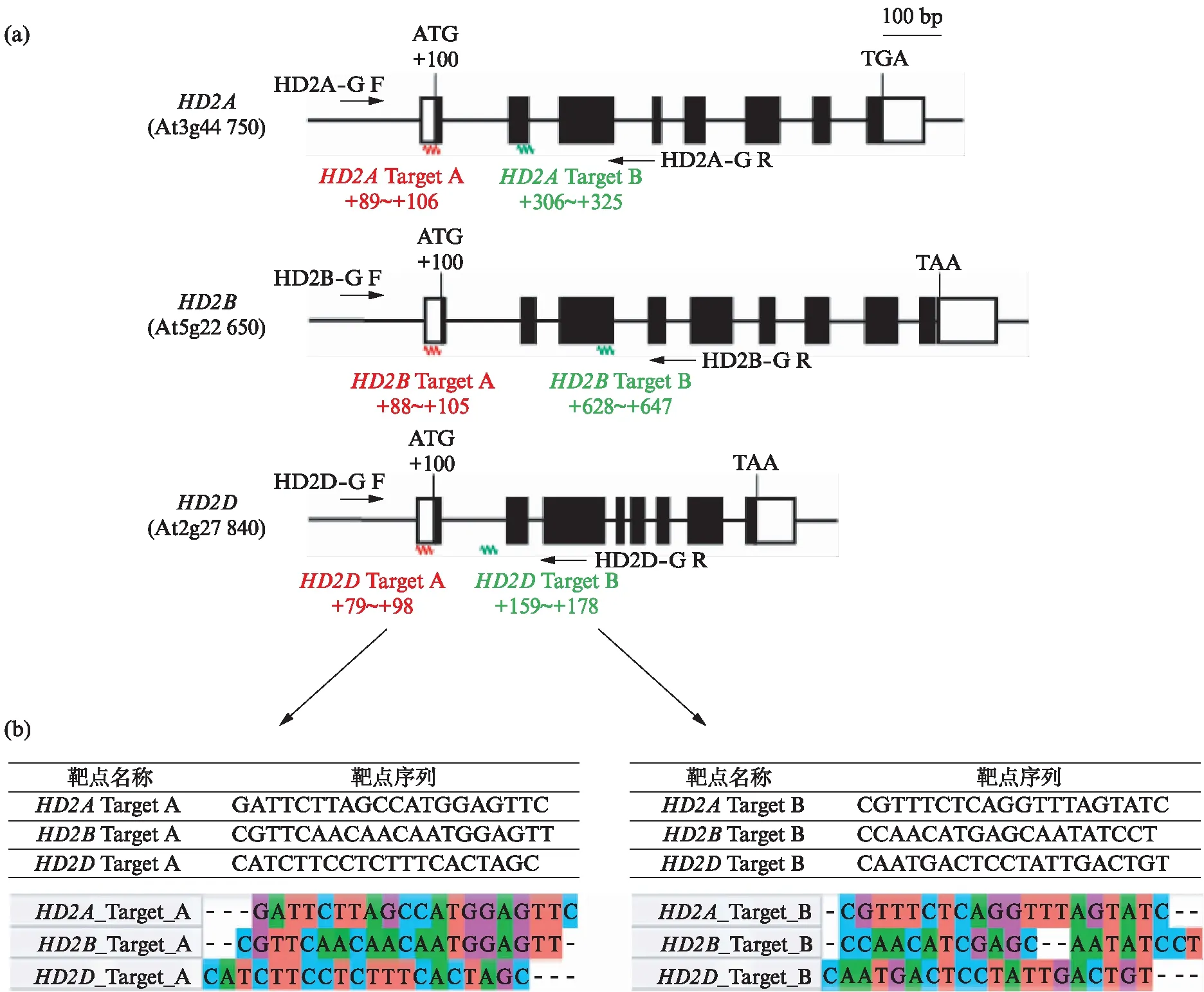
图1 HD2家族基因结构图和编辑位点示意图Fig.1 Gene structure and editing sites of HD2 family(a) HD2家族基因结构图和编辑位点示意图;(b) 箭头所指为靶点序列和靶点通过MEGA软件序列比对后的序列相似性。
载体构建选用GG(Golden Gate)一步克隆策略[20,25]。以基因的靶向序列为模板,分别设计一对gRNA间隔区特异性引物。引物设计原则见图2(a)(见第149页),其中每一个引物5’端的2个碱基(小写字母)为随机添加的保护酶切位点的碱基,3’端的15个碱基为与gRNA退火(正向引物)或与tRNA退火(正向引物)的碱基,5’端的第3~9个碱基为BsaⅠ的酶切位点的特异性序列(斜体大写字母),5’端的第10~21个碱基为基因靶点的特异性序列(大写字母),正向引物为基因靶点的9~20序列,反向引物基因靶点的1~12的反向互补序列,其中正反向引物内有4个碱基的反向互补(粗体大写字母)。引物分别命名为gRNA-HxA-F,gRNA-HxA-R,其中Hx代表HD2基因,A代表靶点A,F代表正向引物,R代表反向引物。载体构建过程如图2(b)所示。以pGTR-sp质粒为模板,用相应的gRNA间隔区特异性正向引物(gRNA-HxA-F)和L5AD5引物、gRNA间隔区特异性反向引物(gRNA-HxA-R)和L3AD5(表1,见第150页)分别扩增得到两条序列,分别用BsaⅠ酶切后,用T4DNA连接酶连接得到PTG产物(序列包括tRNA序列+靶点序列+gRNA序列)。PTG产物用水稀释10倍。以稀释后的PTG产物为模板,用S5AD5和S3AD5引物扩增PTG产物。使用FokⅠ限制性内切酶酶切扩增后的PTG产物,同时用BsaⅠ限制性内切酶酶切Pro: 35S: SpCas9质粒。使用T4DNA连接酶连接两个酶切产物后,使用热激法将其转化大肠杆菌E.coliDH5α感受态。在Kan抗性的固体LB培养基上涂板。阳性菌用OsU3和UGW-gRNA引物(表1,见第150页)菌检,将阳性菌摇菌后提取质粒,送测序,构建好的载体分别命名为pHD2A-AC、pHD2A-BC、pHD2B-AC、pHD2B-BC、pHD2D-AC、pHD2D-BC。把测序正确的6个载体分别通过热激的方法转化到农杆菌A.tumefaciensGV3101感受态中,制备成工程菌。

1.3.2 拟南芥的转化
拟南芥的转化采用浸花法[21]。选择合适开花期的野生型拟南芥植株,在转基因24 h前将植株的果荚和开放的小花全部剪掉,保留未开放的小花蕾。将待转染的农杆菌工程菌小摇培育种子液,随后1∶200(体积比)大摇至OD600=1.3~1.7,离心收集菌体(8 000 r/min,3 min),用缓冲液重悬后,将拟南芥的花序放到含农杆菌的转化介质中浸泡3~5 min进行拟南芥的转染,保湿、避光一天后,置于温室正常生长,两周后再重复转染一次。
1.3.3 拟南芥转基因阳性植株的筛选
阳性苗的筛选采用载体抗性筛选方法。把T0代转基因植株的种子均匀撒在抗性平板(1/2 MS附加25 μg/mL潮霉素(Hygromycin, Hyg)上,筛选T1代阳性苗。
1.3.4 转基因阳性植株基因编辑结果的检测
选用常规基因测序技术检测目的基因的编辑结果。采用CTAB法提取待测植株的DNA,用基因靶点序列检测引物(见表1)分别扩增HD2A,HD2B和HD2D基因的特异序列(引物的位置见图1(a)),琼脂糖凝胶电泳后切下目的条带,纯化后,送生工生物工程(上海)股份有限公司测序。
1.3.5 转基因阳性植株基因表达量的检测和表型观察
表达量的检测选用实时荧光定量PCR(Real-Time PCR)的方法[27]。使用CWBIO公司的植物RNA提取试剂盒提取待测植株叶片的总RNA,使用TaKaRa公司反转录试剂盒合成cDNA第一链。Real-Time PCR扩增使用TaKaRa公司TB GREEN试剂盒,Actin-R为拟南芥的内参基因,采用BIO-RAD定量PCR仪进行扩增。Real-Time PCR程序为95 ℃(10 min);95 ℃(15 s),60 ℃(1 min),共40个循环,溶解曲线默认系统程序(95 ℃,10 s;60 ℃,1 min;95 ℃,15 s)。根据程序导出CT值数据,利用2-ΔΔCT获得相对表达量数据,每个样本3次生物性重复。Real-Time PCR扩增基因对应引物详见表1。

表1 实验所用引物
1.3.6 RNA二级结构预测方法
RNA二级结构预测在http:∥rna.tbi.univie.ac.at/和http:∥www.unafold.org/mfold/applications/rna-folding-form.php网站预测,预测的序列: 20 bp靶点序列和靶点+gRNA骨架,一共96 bp。其中gRNA骨架序列见表1。
2 结果与分析
2.1 gRNA靶点+gRNA骨架二级结构的预测
为了分析RNA的二级结构对基因的编辑率的影响,我们通过网站(http:∥rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNA fold.cgi)预测了不同靶点上的靶点序列和gRNA骨架的二级结构(如图3(a)~(f)①所示),并通过另一个网站(http:∥www.unafold.org/mfold/applications/rna-folding-form.php)预测(如图3(a)~(f)②所示)。两个网站预测的结果大体一致,排除预测结果因为方法、参数选择不同而改变。从图3(a)可以看出HD2ATarget A+gRNA骨架的二级结构由一段单链RNA、5个茎环、1个内饰环、1个突环、1个多环和2个发夹环组成。如果以HD2ATarget A+gRNA骨架的二级结构为标准,那么HD2ATarget B+gRNA骨架多了1个突环,少了1个发夹环(图3(b))。HD2BTarget A+gRNA骨架少了1个多环和1个发夹环(图3(c))。HD2BTarget B+gRNA骨架少了1个内饰环,多了1个发夹环和2个突环(图3(d))。HD2DTarget A+gRNA骨架少了1个内饰环和1个发夹环,多了1个突环(图3(e))。HD2DTarget B+gRNA骨架的茎环、内饰环、多环和发夹环各少1个(图3(f))。gRNA靶点+gRNA骨架二级结构的最小自由能在-26~-21 kcal/mol之间。

图3 gRNA靶点+gRNA骨架的二级结构预测图Fig.3 Prediction of secondary structure of gRNA target+gRNA scaffold(a) HD2A Target A+gRNA骨架;(b) HD2A Target B+gRNA骨架;(c) HD2B Target A+gRNA骨架;(d) HD2B Target B+gRNA骨架;(e) HD2D Target A+gRNA骨架;(f) HD2D Target B+gRNA骨架(根据最小自由能预测gRNA靶点+gRNA骨架的二级结构。不同颜色表示碱基配对概率: 0%~100%)。
同时对20 bp靶点序列的二级结构进行预测。由于20 bp序列较短,导致不同网站预测结果有差别,以下描述以①图为主。自身存在发卡结构的靶位点没有编辑成功(图4(a),4(f)),编辑成功的靶点没有形成发夹结构(图4(c),4(e))。可以成功编辑的靶位点二级结构最小自由能在-0.64~2.00 kcal/mol区间。最小自由能低于-1.00 kcal/mol不能成功编辑(图4(a),4(f))。

图4 20 bp靶点序列的二级结构预测图Fig.4 Prediction of secondary structure of 20 bp target(a) HD2A Target A;(b) HD2A Target B;(c) HD2B Target A;(d) HD2B Target B;(e) HD2D Target A;(f) HD2D Target B(根据最小自由能预测可能性最高的二级结构。不同颜色表示碱基配对概率: 0%~100%)。
2.2 靶位点编辑的统计分析
将工程菌转入拟南芥,收获转基因T1代种子后,进一步的筛选获得T1代的阳性苗。针对HD2A基因的编辑,共获得了41株阳性苗,其中靶点A有23株,靶点B有18株;针对HD2B基因的编辑,共获得了38株阳性苗,其中靶点A有17株,靶点B有21株;针对HD2D基因的编辑,共获得了51株阳性苗,其中靶点A有29株,靶点B有22株,统计结果见表2。将获取的全部阳性苗分别提取DNA后,扩增靶位点上下游序列进行基因编辑位点的检测(如图1所示),结果显示: 针对HD2A基因的编辑,所有41株阳性苗的靶位点序列与野生型拟南芥的序列相同,说明均未发生基因编辑,基因的编辑率为0;针对HD2B基因的编辑,只有靶位点A有2株阳性苗的靶位点序列与野生型拟南芥的序列不同,说明基因成功进行了编辑,基因编辑率为11.7%;针对HD2D基因的编辑,也只有靶位点A有8株阳性苗的靶位点序列与野生型拟南芥的序列不同,说明基因也成功进行了编辑,基因编辑率为27.5%,统计结果见表2。

表2 靶点编辑率统计
2.3 靶位点编辑类型的分析
基因编辑时,一般会在PAM位点的上游3个碱基的位点切割目标DNA。为了确定基因被编辑后是否会对基因的功能产生影响,我们对编辑基因的序列进行了进一步的分析。针对HD2B基因靶位点A的2株阳性植株的分析,发现在靶位点第10个碱基的位置插入了一个G碱基(属于ATG前的第3个碱基,如图5(a)所示),2株阳性植株的编辑位点相同;针对HD2D基因靶位点A的8株阳性植株的分析,发现有5种基因编辑类型: 有2株删除了靶位点的第3位的A碱基(属于ATG前的第19个碱基,如图5(b)所示);有1株为靶位点的第2位T碱基的缺失(属于ATG前的第20个碱基,如图5(c)所示);有1株为靶位点的第4~6位AAG 3个碱基的缺失(属于ATG前的第16~18位碱基,如图5(d)所示);有1株为靶位点第2位前插入一个G碱基(属于ATG前的第19位碱基,图5(e)所示);还有1株编辑了两个位点,一个位点为靶点第2位的碱基G替换了碱基T(属于ATG前的第20个碱基),另一个是靶点前的第一个碱基T的删除(属于ATG前的第22个碱基,如图5(f)所示)。HD2B基因靶位点A和HD2D基因靶位点A的5种基因编辑类型都影响了5’UTR的序列。
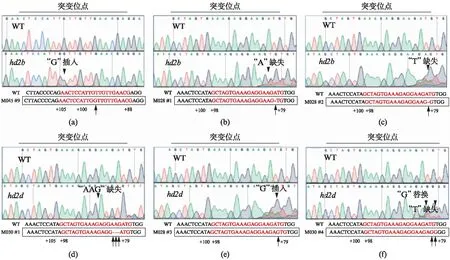
图5 靶位点编辑类型测序结果图Fig.5 Sequences of target site in transgenic plants(a) HD2B Target A中G碱基插入;(b) HD2D Target A中A碱基缺失;(c) HD2D Target A中T碱基缺失;(d) HD2D Target A中AAG碱基缺失;(e) HD2D Target A中G碱基插入;(f) HD2D Target A中G替换T碱基和缺失T碱基(上述都是反向链的测序结果)
2.4 基因编辑突变体与T-DNA插入突变体植株的比较
2.4.1 表达水平的检测
首先对购买的T-DNA插入突变体进行了鉴定。HD2B和HD2D基因的信息和T-DNA插入的位置如图6所示。HD2B基因T-DNA插入在启动子位置,产生的突变体命名为hd2b-1(图6(a));HD2D基因T-DNA也插入在启动子位置,产生的突变体命名为hd2d-1(图6(b))。
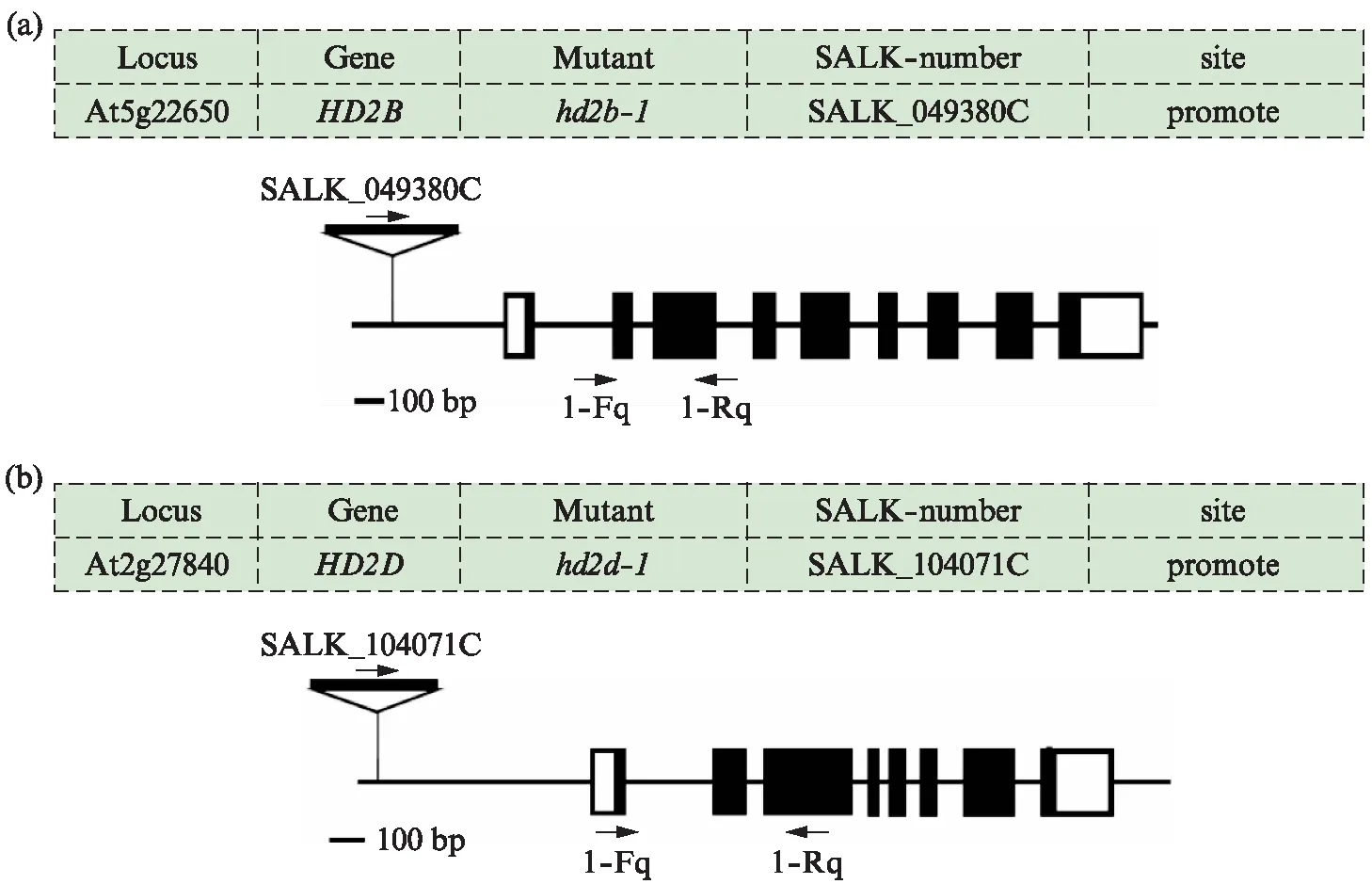
图6 HD2基因T-DNA插入示意图Fig.6 T-DNA insertion of HD2 gene(a) HD2B基因T-DNA插入结构图;(b) HD2D基因T-DNA插入结构图.
采用Real-Time PCR对T-DNA插入突变体和相应的基因编辑突变体的相应基因的表达量进行了检测,T-DNA插入突变体引物的位置如图6所示,引物序列见表1。野生型Col-0的HD2B基因表达量作为对照,hd2b-1T-DNA插入突变体的相对表达量约为0.6,HD2BTarget A中G碱基插入T2代转基因植株命名为hd2b-2,相对表达量为0.9(图7(a));野生型Col-0的HD2D基因表达量作为对照,hd2d-1T-DNA插入突变体的相对表达量约为0.5,HD2DTarget A中A碱基缺失T2代转基因植株命名为hd2d-2,相对表达量为0.8(图7(b))。
2.4.2 叶片表型的观察
取生长约30天的T-DNA插入突变体hd2b-1和hd2d-1以及CRISPR编辑的转基因植株hd2b-2和hd2d-2用于叶片的观察。T-DNA突变体hd2b-1的莲座叶平均数为13,CRISPR编辑的突变体植株hd2b-2的莲座叶平均数为13.8(图7(d))。hd2b-1和hd2b-2叶片长宽比稍微高于Col-0(图7(d))。T-DNA突变体hd2d-1的莲座叶平均数为12.5,CRISPR编辑突变体hd2d-2的莲座叶平均数为13。hd2d-1和hd2d-2叶片长宽比也稍微高于Col-0(图7(d))。

图7 基因编辑突变体与T-DNA插入突变体植株的比较Fig.7 Comparison between gene editing mutants and T-DNA insertion mutants(a) hd2b-1 T-DNA插入和hd2b-2 CRISPR编辑植株中HD2B表达量;(b) hd2d-1 T-DNA插入和hd2d-2 CRISPR编辑植株中HD2D表达量;(c) hd2b-1 T-DNA插入突变体和hd2b-2 CRISPR编辑转基因植株莲座叶;hd2d-1 T-DNA插入突变体和hd2d-2 CRISPR编辑转基因植株莲座叶;(d) 莲座叶数量统计和叶片上宽比统计。每种类型植株统计20株以上,*表示P<0.05(Student t-test),差异显著。此实验重复3次以上,并且结果类似。
3 讨 论
CRISPR/Cas9用于基因组编辑已被广泛应用,该系统在实际应用中主要包含两部分,Cas9蛋白质以及gRNA。gRNA靶位点的正确选择是保证Cas9在特定位点切割DNA的关键。以往的研究主要是依赖在线软件预测的gRNA靶位点,但软件预测出评分最高gRNA不能代表有较高的编辑率,是否能够被正确的编辑需要转基因后进行鉴定,这样往往带有一定的盲目性和滞后性。近期有研究者采用在线软件设计并结合体外酶切检测的方法可大大提高基因的编辑率[17]。
因本文所得的基因编辑阳性株个体数目较少,所得为初步结论。本研究通过对拟南芥HD2家族的3个基因进行CRISPR/Cas9编辑。每个基因各设计2个不同的靶点,不同的靶点会有不同的gRNA接头二级结构。我们发现不同靶点+gRNA骨架的二级结构会有不同的编辑率,当二级结构由一段单链RNA、5个茎环、2个突环和3个发夹环组成的时候,编辑率最高(图3(e))。当gRNA靶点+gRNA骨架的二级结构由一段单链RNA、5个茎环、1个内饰环、1个突环和3个发夹环组成时(图3(c)),CRISPR/Cas9也能成功编辑,但编辑率低,为11.7%。当gRNA靶点+gRNA骨架的二级结构组成为其他类型的时候,CRISPR/Cas9的编辑率为0。此外,20 bp靶点如果自身会形成发夹结构也会影响编辑率(图4(a),4(f))。说明gRNA接头二级结构对以tRNA为策略设计gRNA的CRISPR/Cas9工具极为重要。除了二级结构外,不同基因的染色质开放水平不同可能也会影响基因编辑的效率[28-29],这可能是靶点A更容易编辑成功的原因,后续需要结合ATAC-seq进一步分析[30]。T-DNA插入系中该基因表达水平的检测与编辑个体的检测作比较的目的是让读者除了以Col-0作对照外,还能以T-DNA插入系作对照。HD2B和HD2D的T-DNA插入系的插入位置都在启动子上,而CRISPR/Cas9成功编辑株系的位点在5’UTR上。5’UTR可能影响了mRNA的稳定性[31]。
通过对转基因植株中HD2目的基因测序、Realtime PCR检测和叶片的表型分析,发现特定gRNA接头二级结构的CRISPR/Cas9载体成功编辑了HD2家族的两个基因。说明特定gRNA接头二级结构有助于植物体内RNase P和RNase Z识别tRNA。如果没有相应的gRNA接头二级结构,植物体内部RNase P和RNase Z无法识别,相当于只是转了一个带有Hyg抗性的空载体到野生型中。我们的研究有助于以tRNA为策略设计gRNA的CRISPR/Cas9工具的靶点选择,帮助科研人员选择合适的gRNA接头二级结构,从而有效地对目的基因进行基因编辑,提高CRISPR/Cas9编辑率。
