盐度对疏水改性聚丙烯酰胺吸附行为的影响
2023-01-07邹文杰徐瑞景
邹文杰,饶 博,赵 伟,徐瑞景,王 廷
北京科技大学土木与资源工程学院,北京 100083
选择性絮凝分选是针对微细粒矿物颗粒质量小、难以克服矿化能垒实现有效分选的缺陷而添加的一种有选择性作用的絮凝剂,以选择性增大其中一种组分的颗粒行为尺寸,然后通过沉降或常规浮选将絮体分离的分选方法[1-2],是微细粒矿物高效分选的重要手段之一[3].但是对于复杂的微细粒矿物浮选,现有常用絮凝剂聚丙烯酰胺的絮凝选择性差的问题突出,工业上难以达到好的分选效果.疏水改性聚丙烯酰胺在石油开采等领域受到极大关注,Evani 和Rose[4]于1987 年首次提出了疏水改性聚合物的概念,通过对常规聚丙烯酰胺或部分水解聚丙烯酰胺的主链进行化学改性或与疏水单体共聚,引入极少量疏水基(摩尔分数一般低于2%)而合成.疏水基之间的疏水作用、氢键、静电以及范德华力等相互作用产生分子间或分子内的缔合作用,具有较好的抗剪切、抗盐和抗温的性能,因而在石油开采领域得到应用.Lu 和Huang[5]通过自由基聚合法合成了疏水链长度为C18 的阳离子型疏水单体的疏水改性聚丙烯酰胺,其在黏土矿物表面吸附研究表明,在低质量浓度下(平衡质量浓度小于220 mg·L-1),疏水链是不与黏土表面相互作用的,质量浓度增大时(平衡质量浓度大于310 mg·L-1),疏水链的缔合作用使黏土矿物的表面倾向于多分子层吸附,导致吸附量急剧增加.柳建新等[6]研究了以十六烷基二甲基烯丙基氯化铵为疏水单体改性的聚丙烯酰胺在石英砂上的吸附,研究表明随着疏水基含量的增加,最大吸附量先增加后降低.岳钦艳等[7]采用水溶液自由基胶束聚合法以丙烯酸丁酯为疏水单体的疏水改性聚丙烯酰胺,采用疏水单体含量不同的疏水改性聚丙烯酰胺处理含油废水,取得了优异的除油效果.王红等[8]采用水溶液共聚法制备了以甲基丙烯酸甲酯和丙烯酸丁酯为疏水单体的两种疏水改性聚丙烯酰胺,与聚丙烯酰胺相比,两者对云南某褐铁矿尾矿浆的絮凝效果增强,但对云南某氧化矿尾矿的絮凝效果大幅降低.曹绪龙等[9]发现结构不同的疏水改性聚丙烯酰胺应用在水处理和尾矿处理等方面表现出不同的性能.但在矿物加工领域,聚丙烯酰胺的疏水改性研究尚未引起足够的重视.微细粒矿物分选中,目的矿物在捕收剂作用下表面具有疏水性,脉石矿物表面亲水性强,因此,为强化聚丙烯酰胺对疏水颗粒的选择性絮凝作用,在其主链上引入少量疏水基团进行疏水化改进以适应微细矿物高选择性分选,对于推动选择性絮凝浮选技术的发展具有十分重要的理论意义和实践价值.此外,分选过程中选矿废水处理后需要循环利用,水系统中各种离子反复积累造成循环水水质复杂[10-11],循环水中阳离子对絮凝剂作用的影响也尚不明确.耗散型石英晶体微天平(QCM-D)可用于研究高分子在固-液界面的原位吸附行为以及吸附层的结构[12-15],本文在实验室内合成了疏水改性聚丙烯酰胺(HMPAM),选取了典型亲疏水矿物表面,通过QCM-D 分析了K+及Ca2+浓度对分散剂六偏磷酸钠及HMPAM 在亲疏水矿物表面原位吸附行为的影响,并使用激光粒度分析仪探究不同盐度下吸附SHMP 和HMPAM 后硅微粉及OTS-硅微粉的粒径分布以验证该絮凝剂的絮凝选择性.
1 试验部分
1.1 试验材料
用于合成疏水性单体C16DMAAC 的N,N-二甲基十六胺为分析纯,购自东京化成工业株式会社,单体丙烯酸(AA)和丙烯酰胺(AM)均购自西陇科技有限公司,引发剂过硫酸铵(NH4)2S2O8和亚硫酸氢钠(NaHSO3)均为分析纯,乙醇、丙酮、3-烯丙基氯(纯度≥98.5%)、氢氧化钠(NaOH)、十二烷基硫酸钠(SDS)和十八烷基三氯硅烷(OTS,纯度95%)均购自上海阿拉丁生物化学技术有限公司.
疏水性阳离子单体C16DMAAC 和疏水改性聚丙烯酰胺HMPAM 的合成方法在文献中有详细报道[16-17].即以(NH4)2S2O8与NaHSO3为引发剂,摩尔比为84.7∶15∶0.3 的AM/AA/C16DMAAC 单体采用水溶液聚合法合成疏水改性聚丙烯酰胺HMPAM.所合成HMPAM 的黏均分子量为2.46×106g·mol-1.
1.2 疏水化表面的制备及QCM-D 试验方法
首先将SiO2芯片浸泡在质量分数为2%的SDS溶液中不少于30 min,用超纯水反复冲洗后采用高纯氮气吹干,将芯片置于紫外臭氧仪中10 min.然后用气相沉积法将OTS 沉积在SiO2芯片表面,经测定,清洗后的SiO2表面接触角为20°,改性后的OTS-SiO2表面接触角为65.4°.
石英晶体芯片可测量纳米级沉积或吸附物质质量变化,当其芯片厚度(tq)是诱导波的半波长的奇数倍时,芯片产生共振.石英晶体表面质量增加时,其谐振频率降低,且降低的幅度与增加的质量成正比[18-19].
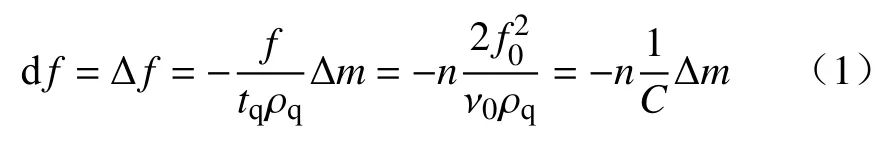
其中:C是常数(C=17.7 ng·cm-1·Hz-1);n是泛频数(n=1,3,···);tq是芯片厚度,cm;f是谐振频率,Hz;f0是芯片固有的谐振频率,Hz;Δm是工作电极上沉积物的质量改变,g;ρq是石英晶体的密度,kg·m-3;ν0是石英晶体的剪切模量,Pa.Sauerbrey 方程仅适合于真空或空气中的刚性薄膜(吸附的薄膜完美贴合在芯片表面).在QCM-D 系统中,芯片能量的耗散(D)可用来描述吸附薄膜的黏弹性,其表达式为:
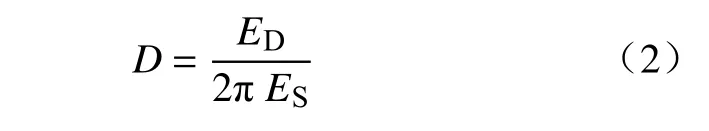
式中:ED表示晶体在一次振荡周期中能量损耗;ES表示晶体在一次振荡周期中存储的全部能量[20].通过两种基本模型(Maxwell 模型和Voigt 模型)中的剪切黏度系数和剪切弹性模量来测量耗散因子,体系中液体的密度和黏度分别为996 kg·m-1和0.001 Pa·s[21].
试验采用瑞典Q-Sense 的QCM-D E1 系统.首先室温下配置质量分数为0.1%的HMPAM 溶液,磁力搅拌器缓慢搅拌时间约为10 h,配置KCl浓度分别为10 mmol·L-1和100 mmol·L-1以 及CaCl2浓度分别为1 mmol·L-1和10 mmol·L-1的背景电解质溶液以及浓度为10 mmol·L-1的SHMP 溶液,后续根据需要稀释为SHMP 和HMPAM 试验溶液.将KCl 或者CaCl2背景溶液泵入QCM-D 模块以记录基线,待基线平稳后将频率f和耗散值D定义为0;然后泵入不同背景溶液的SHMP 溶液直到获得稳定信号,随后通入KCl 或者CaCl2背景溶液进行冲洗,稳定后继续通入不同背景溶液的100 mg·L-1的HMPAM 溶液,获得稳定的频率信号后通入KCl 或者CaCl2溶液,以去除被捕获或松散结合的聚合物[22],实验温度设置为20±0.02 ℃,流量为100 μL·min-1.通过Q-sense Dfind 软件对Δf和芯片能量的耗散变化(ΔD)曲线进行分析.
1.3 OTS-硅微粉的制备及激光粒度分析仪实验方法
取硅片及硅微粉浸泡在质量分数为2%的SDS溶液中不少于30 min,用超纯水反复冲洗,过滤后沉淀物放入真空干燥箱30 ℃烘干4 h;将OTS 与甲苯混合配置成5 mmol·L-1OTS/甲苯溶液,干燥后的硅片及硅微粉分别放入上述溶液浸泡6 min,先后用甲苯溶液、超纯水反复清洗,放入真空干燥箱30 ℃烘干4 h 后得到OTS-硅片及OTS-硅微粉.OTS-硅片表面去离子水的接触角为63.3°.
采用欧美克LS-POP(9)激光粒度分析仪测试硅微粉及OTS-硅微粉的粒径分布.先用去离子水配制30 g·L-1矿浆,超声分散3 min,向激光粒度分析仪的进样器中加入300 mL 一定浓度的KCl 或CaCl2溶液,将超声分散后的矿浆逐步加入进样器至激光粒度分析仪的遮光比达到13%~20%范围,调节进样蠕动泵在合适的流动速度下搅拌循环10 min后,采集空白对照组粒度分布数据,然后加入SHMP溶液使其浓度达到1 mmol·L-1,搅拌循环10 min 后开始采集粒度分布数据,然后加入HMPAM 溶液至其质量浓度达到100 mg·L-1,搅拌循环10 min 后开始采集粒度分布数据,当粒径不再变化,取该值为药剂作用后絮体粒径分布结果.
2 试验结果与讨论
采用QCM-D 测定了在不同盐度的背景溶液(10 mmol·L-1KCl、100 mmol·L-1KCl、1 mmol·L-1CaCl2和10 mmol·L-1CaCl2)下,1 mmol·L-1SHMP 和100 mg·L-1HMPAM 分别在OTS-SiO2和SiO2表面的原位吸附行为,可获得频率(f)和耗散值(D)随时间的变化.
2.1 不同盐度下SHMP和HMPAM在疏水化表面的吸附行为
图1 为分别在10 mmol·L-1KCl、100 mmol·L-1KCl的背景溶液中,1 mmol·L-1SHPM 和100 mg·L-1HMPAM 在疏水OTS-SiO2表面的吸附行为.由图1(a)可见,通入10 mmol·L-1KCl 背景溶液基线稳定归零后,泵入1 mmol·L-1SHMP、10 mmol·L-1KCl 溶液,Δf、ΔD保持在基线上,用10 mmol·L-1KCl 冲洗后,Δf、ΔD未发生变化,表明未测得SHMP 吸附层.随后泵入100 mg·L-1HMPAM 溶液后,Δf急剧下降至-15.5 Hz,ΔD上升至8.4×10-6,说明HMPAM 在疏水表面吸附形成了一个相对厚且松散的吸附层.
当背景溶液KCl 浓度增加到100 mmol·L-1时,如图1(b)所示,通入1 mmol·L-1SHMP 溶液后,Δf下降至-2.0 Hz,ΔD上升为0.78×10-6,用100 mmol·L-1KCl 溶液冲洗后,Δf、ΔD稳定在零基线位置,表明SHMP 在OTS-SiO2疏水表面的吸附是可逆的,该吸附层可被背景溶液冲洗掉.随后泵入100 mg·L-1HMPAM 溶液后,Δf下降至-15.8 Hz,ΔD为6×10-6,有所下降,即100 mmol·L-1KCl 溶液中HMPAM 在疏水化表面也形成了一个相对厚的吸附层,该吸附层较在10 mmol·L-1KCl 背景溶液下所形成的HMPAM吸附层更加密实性.以上研究表明,在10 mmol·L-1KCl 和100 mmol·L-1KCl背景溶液中,SHMP 并 不会吸附在OTS-SiO2疏水表面上,也不会影响HMPAM在疏水矿物表面的吸附,10~100 mmol·L-1KCl 范围内,HMPAM 吸附层厚度略有增加,吸附层的耗散性减弱.
图1 1 mmol·L-1 SHMP 和100 mg·L-1 HMPAM 在OTS-SiO2 表面的吸附行为.(a)10 mmol·L-1 KCl;(b)100 mmol·L-1 KClFig.1 Effect of 1 mmol·L-1 SHMP and 100 mg·L-1 HMPAM on adsorption behavior of OTS-SiO2: (a) in 10 mmol·L-1 KCl;(b) in 100 mmol·L-1 KCl
二价阳离子也会在选矿的水系统中循环和积累,图2 为背景溶液分别为1 mmol·L-1和10 mmol·L-1CaCl2时,1 mmol·L-1SHPM 和100 mg·L-1HMPAM 在OTS-SiO2表面的吸附行为.如图2(a)所示,泵入1 mmol·L-1SHMP 溶液后,Δf下降到-1.9 Hz,ΔD上升到2.3×10-6,用1 mmol·L-1CaCl2冲洗后,Δf、ΔD回归到基线位置,则未测得SHPM 在OTS-SiO2表面的吸附层;随后泵入100 mg·L-1HMPAM 溶液,并用背景溶液冲洗达到稳定后,Δf下降至-12.5 Hz,ΔD降低稳定在4.6×10-6,在OTS-SiO2疏水表面形成了HMPAM 吸附层.
图2 1 mmol·L-1 SHMP 和100 mg·L-1 HMPAM 在OTS-SiO2 表面的吸附行为.(a) 1 mmol·L-1 CaCl2;(b) 10 mmol·L-1 CaCl2Fig.2 Effect of 1 mmol·L-1 SHMP and 100 mg·L-1 HMPAM on adsorption behavior of OTS-SiO2: (a) in 1 mmol·L-1 CaCl2;(b) in 100 mmol·L-1 CaCl2
图2(b)为在10 mmol·L-1CaCl2背景溶液下,加入1 mmol·L-1SHMP溶液后,Δf下降到-3.2 Hz,ΔD上升到5.5×10-6,有较为松散的吸附层在该疏水化表面生成;用10 mmol·L-1CaCl2溶液冲洗后,Δf下降到-5.3 Hz,ΔD下降到4.0×10-6,则在疏水矿物表面继续形成了较为致密的沉积层,该沉积层可能为固态Ca(H2PO4)2及其水合物[23];在通入100 mg·L-1HMPAM 溶液后,Δf下降至-23.6 Hz,ΔD上升至11.6×10-6,该吸附层的Δf为-18.3 Hz,ΔD为7.6×10-6,则虽然有固体沉淀沉积在疏水表面,但并未抑制HMPAM在疏水表面的吸附,且吸附量较在KCl 背景溶液下有所增加,这是由于固体沉淀并未覆盖全部的疏水表面,Ca2+可使聚合物更加卷曲,在表面吸附位点增多而导致吸附量增加[24-25].
2.2 不同盐度下SHMP 和HMPAM 在SiO2 表 面的吸附行为
图3 为背景溶液分别为100 mmol·L-1KCl、10 mmol·L-1CaCl2中1 mmol·L-1SHMP 和100 mg·L-1HMPAM 在SiO2表面的吸附行为.由图3(a),通入100 mmol·L-1KCl 为背景溶液的1 mmol·L-1SHMP,Δf降低至-0.7 Hz,ΔD升高至0.2×10-6,用100 mmol·L-1KCl 溶液冲洗后,Δf上升到-0.2 Hz,ΔD回归基线位置,则在SiO2表面上吸附了薄而刚性的SHMP吸附层;随后泵入100 mg·L-1HMPAM 溶液后,Δf、ΔD未发生变化,说明在二氧化硅表面上吸附的SHMP 抑制了HMPAM 的吸附.
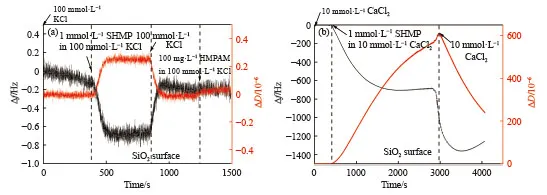
图3 1 mmol·L-1 SHMP 和100 mg·L-1 HMPAM 在SiO2 表面的吸附行为.(a) 100 mmol·L-1 KCl;(b) 10 mmol·L-1 CaCl2Fig.3 Effect of 1 mmol·L-1 SHMP and 100 mg·L-1 HMPAM on adsorption behavior of SiO2: (a) in 100 mmol·L-1 KCl;(b) in 10 mmol·L-1 CaCl2
由图3(b)在10 mmol·L-1CaCl2背景溶液下,泵入1 mmol·L-1SHMP 溶液后,Δf降低至-690 Hz,ΔD升高至约605×10-6,该吸附量较在疏水化表面高出2 个数量级,且是耗散的吸附层;随后通入10 mmol·L-1CaCl2溶液冲洗后,Δf继续下降至-1250 Hz,ΔD值也逐渐下降至240×10-6,说明继续通入10 mmol·L-1CaCl2冲洗过程中形成了较为致密的吸附层,已有研究证明该沉积层可能为固态Ca(H2PO4)2及其水合物,前期研究表明该吸附层可抑制HMPAM 在SiO2的吸附.图4 为在10 mmol·L-1CaCl2溶液环境下,SHMP 与SiO2表面反应的SEM/EDS 图像.

图4 1 mmol·L-1 SHMP 在10 mmol·L-1 CaCl2 溶液环境下SiO2 基底表面的SEM/EDS 图像Fig.4 SEM/EDS imagaes of silica surface with effect of 1 mmol·L-1 SHMP in 10 mmol·L-1 CaCl2 solution
综上,对于OTS-SiO2为典型代表的疏水化表面,在10 mmol·L-1KCl、100 mmol·L-1KCl 和1 mmol·L-1CaCl2背景溶液中,SHMP 均未在OTS-SiO2疏水表面测得吸附层,HMPAM 在疏水矿物表面形成吸附层.10 mmol·L-1CaCl2与SHMP 可生成固体沉淀沉积在疏水化表面,但并未抑制HMPAM 的吸附,因吸附层构象重构吸附量反而增加.对于SiO2为典型代表的亲水表面,100 mmol·L-1KCl 和10 mmol·L-1CaCl2背景溶液下,SHMP 在SiO2表面形成的吸附层可以抑制HMPAM 的吸附.因此,在所考察的KCl和CaCl2浓度范围内,在SHMP 的作用下,HMPAM在亲疏水表面呈现了较好的吸附选择性.
2.3 不同盐度下SHMP和HMPAM对硅微粉及OTS-硅微粉粒径分布的影响
图5 为在10 mmol·L-1KCl 和100 mmol·L-1KCl背景溶液下,SHMP 和HMPAM 作用前后OTS-硅微粉的表观粒径分布曲线.SHMP 作用后的OTS-硅微粉的粒度分布峰形整体变化不大,与空白对照组的粒度分布基本重合;HMPAM 作用后OTS-硅微粉的粒度分布峰形发生了变化且整体向右移动,并且与未加药剂前OTS-硅微粉的粒度分布重合较少,相对比较独立,说明先后在SHMP、HMPAM作用下OTS-硅微粉发生絮凝产生了絮体,对于疏水化的硅微粉表面,SHMP 并未抑制HMPAM 的絮凝作用.
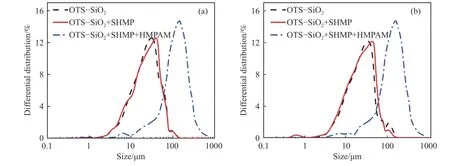
图5 1 mmol·L-1 SHMP 和100 mg·L-1 HMPAM 作用前后OTS-硅微粉的表观粒径分布.(a) 10 mmol·L-1 KCl;(b) 100 mmol·L-1 KClFig.5 Effect of 1 mmol·L-1 SHMP and 100 mg·L-1 HMPAM on the floc size distribution of OTS-silica powder: (a) in 10 mmol·L-1 KCl;(b) in 100 mmol·L-1 KCl
图6 为在1 mmol·L-1CaCl2和10 mmol·L-1CaCl2背景溶液下,SHMP 和HMPAM 作用前后OTS-硅微粉的表观粒径分布曲线.在1 mmol·L-1CaCl2背景溶液下,SHMP 作用后的OTS-硅微粉的粒度分布峰形与空白对照组的粒度分布峰形有较大重合;但在10 mmol·L-1CaCl2背景溶液中,SHMP 作用后的OTS-硅微粉的粒度分布峰形基本未发生变化但整体向右移动,说明在10 mmol·L-1CaCl2的高盐度和SHMP 的作用下OTS-硅微粉的絮体粒径增大,该粒径增大推测是由于有沉淀生成沉积在颗粒表面所致;随后通入HMPAM 溶液后,OTS-硅微粉的粒度分布峰形发生变化且再次向右移动,说明HMPAM 作用下OTS-硅微粉发生絮凝使得絮体粒径增大.
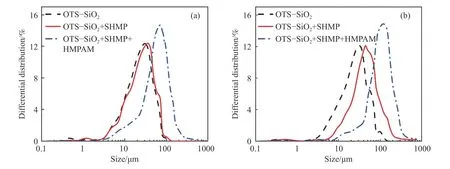
图6 1 mmol·L-1 SHMP 和100 mg·L-1 HMPAM 作用前后OTS-硅微粉的表观粒径分布.(a)1 mmol·L-1 CaCl2;(b) 10 mmol·L-1 CaCl2Fig.6 Effect of 1 mmol·L-1 SHMP and 100 mg·L-1 HMPAM on floc size distribution of OTS-silica powder: (a) in 1 mmol·L-1 CaCl2;(b)10 mmol·L-1 CaCl2
图7 为在100 mmol·L-1KCl 和10 mmol·L-1CaCl2背景溶液下,SHMP 和HMPAM 作用前后硅微粉表观粒径分布曲线.SHMP 作用后硅微粉的粒度分布整体向右移动,峰形变化不大;HMPAM 作用后硅微粉的粒度分布峰形整体没有变化,并且与作用前样品的粒度分布峰形重合较多,即硅微粉未发生絮凝,说明SHMP 阻止了HMPAM 絮凝硅微粉.综上,不同盐度下SHMP 和HMPAM 对硅微粉及疏水化硅微粉粒径分布的影响与QCMD 试验结果相一致.
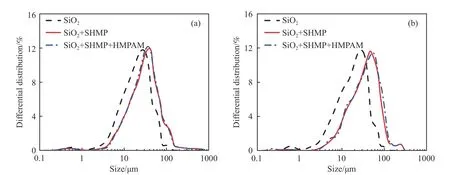
图7 1 mmol·L-1 SHMP 和100 mg·L-1 HMPAM 作用前后硅微粉的表观粒径分布.(a)100 mmol·L-1 KCl;(b)10 mmol·L-1 CaCl2Fig.7 Effect of 1 mmol·L-1 SHMP and 100 mg·L-1 HMPAM on floc size distribution of silica powder: (a) in 100 mmol·L-1 KCl;(b) in 10 mmol·L-1 CaCl2
3 结论
本文将疏水基团C16DMAAC 引入PAM 分子链中合成了疏水改性聚丙烯酰胺HMPAM,使用QCM-D 系统地研究了不同盐度的背景溶液下,SHMP 和HMPAM 在亲水表面SiO2和疏水表面OTS-SiO2的原位吸附行为,并通过对硅微粉及OTS-硅微粉的絮凝行为进行验证.研究表明在所考察的KCl 和CaCl2浓度范围内,在SHMP 的作用下HMPAM 在亲疏水表面呈现了较好的絮凝选择性.在10 mmol·L-1KCL、100 mmol·L-1KCl 和1 mmol·L-1CaCl2溶液中,SHPM 都无法吸附在OTSSiO2疏水表面,10 mmol·L-1CaCl2与SHMP 可生成Ca(H2PO4)2及其水合物等固体沉淀沉积在疏水化表面,均未抑制HMPAM 的吸附,吸附量反而增加;对于SiO2表面,100 mmol·L-1KCl 和10 mmol·L-1CaCl2背景溶液下,SHMP 在SiO2表面形成的吸附层均抑制了HMPAM 的吸附,本研究进一步明确了分选过程中选矿循环水系统中阳离子对絮凝剂作用的影响.
