电化学合金-去合金法制备铂纳米粒子电极检测尿液中草酸
2023-01-05王焕焕郭禹红
苏 敏,王焕焕,郭禹红,金 君,于 浩,2*
(1.延安大学 化学与化工学院,陕西 延安 716000;2.陕西省化学反应工程重点实验室,陕西 延安 716000)
草酸(OA),又名乙二酸,广泛分布于植物、动物和真菌体中。OA能参与细胞Ca2+、pH值和渗透压等的调节[1]。尽管OA有重要生理功能,但过量摄入会增加人体患肾结石、炎症、高草酸尿症、尿毒症等疾病的风险,严重的甚至会引起OA急性中毒[1]。人体内OA经血液循环后会以尿的形式排出体外。因此,建立尿液中OA含量的检测方法具有重要意义。目前,尿液中OA的检测方法主要包括光谱法[2]、色谱法[1,3]、毛细管电泳法[4]和电化学法[5-6]等。在这些方法中,电化学法因具有仪器简单、灵敏度高等优点在尿液分析中受到了广泛的重视。
电化学检测OA主要依据其可被电化学氧化的原理进行。但OA在传统电极上氧化时的过电位高,需对电极表面进行必要的修饰以提高方法的灵敏度和选择性。金属纳米材料如Pd[5-9]、Pt[10-12]、Ag[13]、Ag/AgCl[14]和碳化钨(WC2)[15],以及碳基纳米材料如硼掺杂钻石[16]和酸化碳纳米管[17]等已被用作电极修饰材料以提高电化学检测OA的分析性能。贵金属Pt以其优异的催化性能及耐腐蚀性在电化学传感领域得到了广泛的应用[10-12]。但Pt基材料的电催化活性与其形貌及粒径密切相关,为了进一步提高催化活性,不同结构和形貌的Pt纳米材料被相继制备并用于OA的电化学传感[18-21]。在各种Pt基纳米材料中,多孔结构Pt纳米材料具有高可接触性、大比表面积及相互连接的孔隙,表现出优异的离子/电子传输性能及扩散性能,在电化学传感领域中引起了人们广泛的研究兴趣[22-23]。目前,模板法、去合金法及自组装法等已用于制备多孔结构Pt纳米材料[22]。其中电化学去合金技术以成本低、结构与形貌可控等特点在多孔结构纳米材料的制备中受到了重视[24]。但传统电化学去合金技术制备多孔结构纳米材料修饰电极时程序复杂[25]。最近研究表明,可采用电化学方法直接将金属合金共沉积于电极表面,再电化学去合金得到多孔金属纳米材料。如Beluomini等[26]先采用电化学方法将Pt-Cu合金膜沉积于基础电极表面,然后采用循环伏安法溶掉其中的Cu,得到一种多孔结构Pt纳米粒子修饰电极,这种电化学合金-去合金技术大大简化了多孔结构金属纳米材料修饰电极的制备程序。
本文以Bi为活泼金属,采用电化学合金-去合金法制备了多孔结构Pt纳米粒子修饰电极。结果表明,多孔结构能有效增大铂纳米粒子的活性面积,同时也有利于充分暴露电极表面的活性位点和待测物质向电极表面的传质,显示出对电氧化草酸的强催化活性。在此基础上,采用安培法定量测定了人尿液中的OA含量。
1 实验部分
1.1 仪器与试剂
CHI660D电化学工作站(上海辰华仪器公司),实验采用三电极系统:以修饰电极为工作电极,铂柱电极为辅助电极(Pt),饱和甘汞电极为参比电极(SCE)。SU5000热场发射扫描电子显微镜(日本日立公司,Hitachi Limited)。草酸钠(Na2C2O4,优级纯,天津市科密欧化学试剂厂),氯铂酸钾(K2PtCl6,分析纯,国药集团化学试剂有限公司),硝酸铋(Bi(NO3)3·5H2O,分析纯)、乙二胺四乙酸二钠盐(EDTA,分析纯)购自天津市科密欧化学试剂开发中心,其它试剂均为分析纯,实验用水为二次蒸馏水。
1.2 修饰电极的制备
参考文献制备Pt纳米粒子修饰电极[27]:先将处理好的玻碳电极(GCE)置于3.0 mmol/L Bi(NO3)3+1.0 mmol/L K2PtCl4+3.0 mmol/L EDTA+0.10 mol/L HClO4混合溶液中,在-0.4 V电位下恒电位沉积500 s,制得Pt-Bi合金修饰的玻碳电极(Pt-Bi/GCE)。取出冲洗干净后置于0.10 mol/L HClO4中,在1.2 V下阳极化处理1 000 s,溶去合金中的Bi,即得Pt纳米粒子修饰的玻碳电极(Pt NPs/GCE)。采用相似方法从不含Bi(NO3)3的溶液中制备Pt修饰电极,记为Pt/GCE。
1.3 电极表征及实验方法
电极表征:将制备好的电极置于扫描电镜样品台上记录SEM照片及EDS能谱。将制备好的电极置于0.10 mol/L HClO4中采用循环伏安法扫描至稳定后,记录加入一定浓度OA前后的CV图,研究OA的电化学性质。控制工作电位为1.05 V,向10 mL不断搅拌的0.10 mol/L HClO4溶液中加入一定体积的OA标准溶液和样品溶液,记录I~t曲线,测量电流,绘制标准曲线,用标准曲线法计算样品中OA的浓度及加标回收率。
2 结果与讨论
2.1 修饰电极的制备及表征
本实验以Bi(NO3)3和K2PtCl6为前驱体制备Pt-Bi合金,由于Bi3+离子与Cl-缓慢生成BiOCl沉淀,故向沉 积 液 中 加 入EDTA。图1A是 在1.0 mmol/L K2PtCl6+3.0 mmol/L Bi(NO3)3+3.0 mmol/L EDTA+0.10 mol/L HClO4溶液中记录的CV图,其中-0.329 V处的还原峰为Bi3+的本体还原峰,-0.011 V处的还原峰为PtCl2-6离子的还原峰;阳极扫描时,CV图上-0.019、0.095 V处的氧化峰是Bi的本体溶出峰,而0.491、0.601、0.778 V处的氧化峰来源于合金中Bi的溶出及Pt氧化物的形成。为了证明这一点,记录了只含Bi3+和EDTA的CV图(图1B),-0.411 V处出现Bi3+的本体还原峰,而Bi的溶出峰出现在0.103 V。与Pt-Bi体系相比,Bi3+的还原峰电位更负,这是由于在Pt-Bi体系中,Bi沉积在先析出的Pt颗粒表面,因而过电位降低。其次,在图1B上,除Bi的本体氧化溶出峰外,CV图上无其它氧化峰。而在只含PtCl2-6和EDTA的溶液中,CV图在-0.203 V处出现PtCl2-6的还原峰,当电位进一步负移时,电流迅速增大,这是析出H2所致,阳极1.00 V处的峰是Pt氧化物的峰(图1C)。另外,在Pt-Bi体系中(图1A),低电位区并未出现析出H2时的大电流,这是由于Pt-Bi体系中的Bi沉积在先析出的Pt颗粒表面,从而将Pt颗粒表面覆盖,而Bi对析氢反应有抑制作用所致。以上结果说明在上述溶液中可实现Pt、Bi在电极表面的共沉积,从而得到Bi-Pt合金,且Bi在Pt颗粒表面的沉积会干预Pt粒子的沉积,从而控制Pt颗粒的粒径。进一步电化学溶出Bi后,可得到粒径小且分布均匀的Pt纳米粒子。根据CV行为,本实验选择-0.4 V作为合金沉积时的工作电位,在1.2 V去合金。图1D和E分别为电化学合金及去合金时的计时安培曲线,可观察到二者均在短时间内使电流基本达到稳态,说明Pt-Bi合金被均匀地沉积于电极表面,同时也表明去合金过程均匀稳定,有利于得到分布均匀的Pt粒子[27]。图1F是无Bi(曲线a)和有Bi(曲线b)时制备的Pt纳米粒子修饰电极在0.5 mol/L H2SO4溶液中的CV图。根据CV图上低电位区H的吸脱附峰面积可计算电极的电化学活性面积(ECSA)[28]。经计算,Pt NPs/GCE和Pt/GCE的电化学活性面积分别为0.201、0.112 cm2。表明电化学合金-去合金法制备的Pt粒子具有更大的活性面积。

图1 修饰电极的电化学制备Fig.1 Electrochemical preparation of the modified electrode
图2A、B是电化学去合金前、后Pt-Bi电极表面的扫描电镜图。可以看出,去合金前,电极表面密集分布着Pt-Bi合金颗粒,且大呈立方块状。去除Bi后,电极表面的颗粒密度明显减少,且立方块状结构消失,这是由于去合金时,合金中的Bi被电化学溶出,留下了Pt颗粒所致。另一方面,去合金后颗粒在电极表面分布较均匀,表明采用本法可制备Pt纳米颗粒修饰电极(图2B)。而单独沉积Pt时只能得到团聚严重、颗粒较大的Pt粒子,且分布不均匀(图2C)。图2D是不同电极表面的EDS能谱图。可以看出,Pt-Bi合金在2.055 keV和2.415 keV处分别出现了Pt和Bi元素的特征峰(曲线a),说明采用电化学方法可将Pt-Bi合金沉积于电极表面。去合金后,2.415 keV处Bi元素的特征峰消失(曲线b),说明采用电化学去合金法可将Bi溶出。单独沉积Pt时EDS能谱只有Pt的特征峰,无Bi的特征峰(曲线c)。以上结果说明,采用电化学合金-去合金法可制备Pt纳米结构,且Bi的共沉积干预了Pt的沉积过程,得到了粒径更小、分布更均匀的Pt纳米颗粒,有利于增大电极的有效面积,提高电流响应。
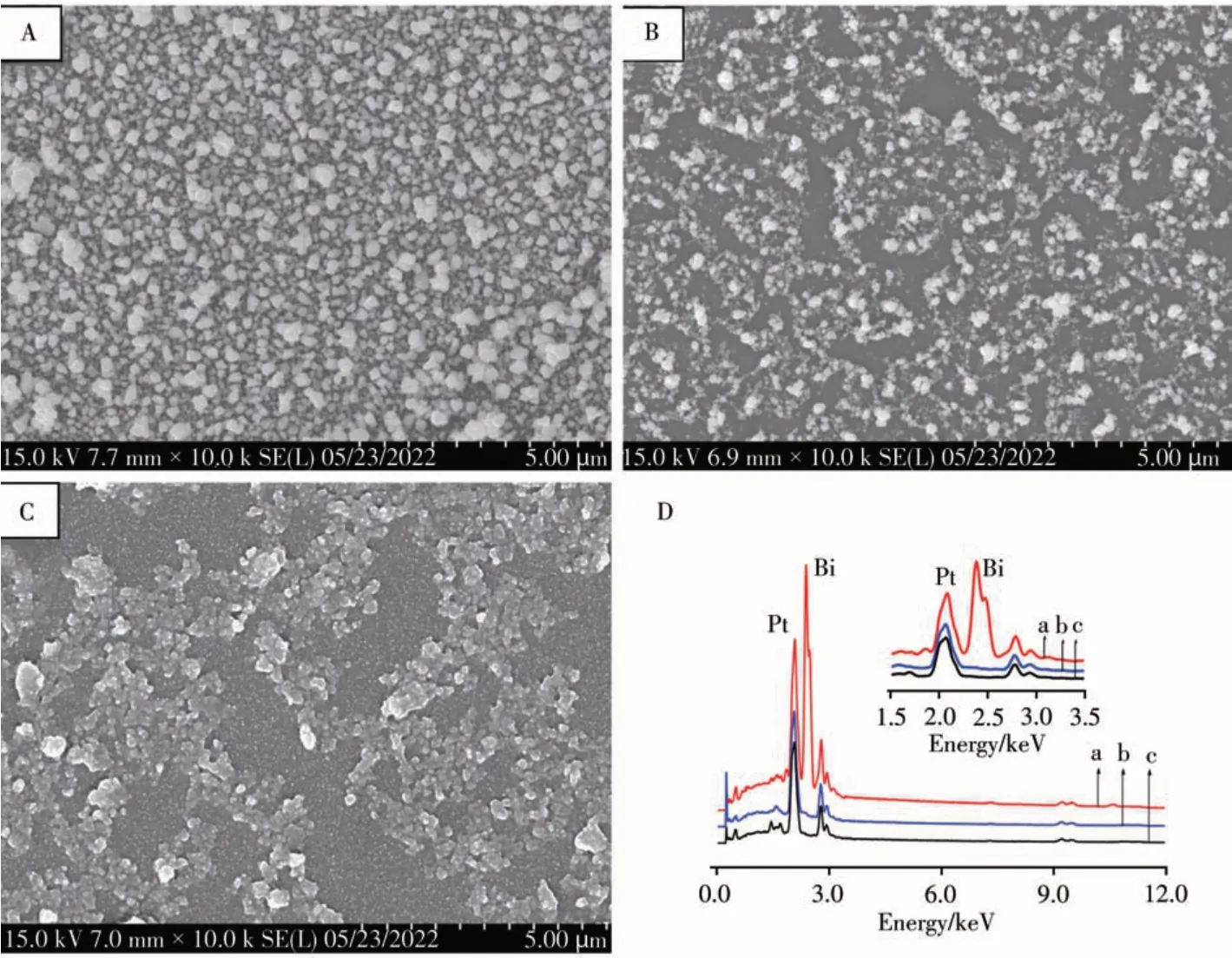
图2 不同电极表面的扫描电镜图(A~C)及EDS能谱图(D)Fig.2 SEM(A-C)and EDS(D)graphs of different electrodes
2.2 修饰电极对草酸的电催化氧化
根据文献报道,OA的电化学氧化更易在强酸性介质中进行[10,29]。同时,OA的电极过程包含OA分子在电极表面的吸附,所以支持电解质阴离子的种类对其电催化活性有影响[29]。分别研究了该电极在HCl、H2SO4和HClO4中对OA的电氧化行为。结果表明,在0.10 mol/L HClO4溶液中该修饰电极对OA的催化氧化活性最高,故以其作为支持电解质溶液。图3为不同电极加入不同浓度OA前、后的CV图。在Pt NPs/GCE上,OA在0.828 V处产生一个尖锐的不可逆氧化峰(图3A),1.0 mmol/L OA的峰电流密度为56.37 μA/cm2,峰电流与OA浓度间线性关系的斜率为57.71 μA/(mmol·L-1·cm-2);而在Pt/GCE上,OA的氧化峰位于0.873 V(图3B),1.0 mmol/L OA的峰电流密度为42.95 μA/cm2,峰电流与OA浓度间线性关系的斜率为44.11 μA/(mmol·L-1·cm-2)。在裸玻碳电极上,OA在1.5 V电位附近产生一个峰形很斜的不可逆氧化峰(图3C)。通过对比,OA在Pt NPs/GCE上具有更低的过电位和更大的电流响应,这是由于采用电化学合金-去合金法制备的Pt NPs具有更小的粒径和更大的活性面积。

图3 不同电极加入不同浓度OA前(a)后(b-d)的CV图Fig.3 CV graphs of different electrodes without(a)and with(b-d)addition of various concentration of OA
采用循环伏安法研究了电位扫描速率的影响(图4A)。在20~150 mV/s范围内,峰电流与扫速的1/2次方成正比(图4B),说明电极过程受OA的扩散控制,且峰电流对数与扫速对数呈线性关系(图4C),斜率为0.48,接近理论值0.50,说明OA在该电极上是一个完全不可逆的阳极过程。采用低扫速时CV图上峰电流10%以内的lgI~E曲线估测电极反应的电子转移数,图4D为扫速为20 mV/s时,0.70~0.75 V范围内的lgI~E线性关系,其斜率为9.79,接近单电子转移不可逆过程的理论值(9.26),说明OA在该电极上的速控步骤是一个单电子转移过程,据此求得电子传递系数α=0.54[7]。

图4 扫描速率对OA在Pt NPs/GCE上电化学行为的影响Fig.4 Effect of scan rate on the electrochemical behavior of OA on Pt NPs/GCE
2.3 电极制备条件的优化
考察了沉积液中Bi3+和PtCl2-4浓度比例(cBi∶cPt)的影响:保持沉积液中PtCl2-4浓度为1.0 mmol/L,改变Bi(NO3)的浓度并使EDTA的总浓度与Bi3+离子浓度相同,配制了一系列不同cBi∶cPt的沉积液,分别制备修饰电极,并记录不同电极加入等量OA前后的CV图。图5A结果显示,当Bi3+浓度在0.5~4.0 mmol/L范围内变化时,随着Bi3+浓度的增大,峰电流先增大后减小,当Bi3+浓度为3.0 mmol/L时,峰电流最大,故选择两种离子浓度比为3∶1。采用相似方法研究了沉积时间的影响,图5B结果显示,在100~1 000 s范围内,随着沉积时间的延长,OA峰电流先增大后减小,当沉积时间为500 s时峰电流最大,故选择合金沉积时间为500 s。当去合金时间为合金沉积时间的2倍时[27],可保证将合金中的Bi完全溶出,这可通过去合金前后的EDS能谱图证明(图2D)。最终选择电极制备条件如下:沉积液组成为1.0 mmol/L K2PtCl6+3.0 mmol/L Bi(NO3)3+3.0 mmol/L EDTA+0.10 mol/L HClO4,合金沉积时间为500 s(-0.4 V),去合金时间为1 000 s(1.2 V)。

图5 沉积液中Bi3+浓度(A)及沉积时间(B)的影响Fig.5 Effects of Bi3+concentration(A)and deposition time(B)on the peak current
2.4 安培法对草酸的测定
2.4.1 工作电位的优化以0.10 mol/L HClO4作为支持电解质溶液,采用安培法记录了不同工作电位下向不断搅拌的支持电解质溶液中连续5次加入10.0 μmol/L OA时的动力学安培响应曲线。结果显示,在0.60~1.20 V范围内,随着工作电位的升高,电流逐渐增大,至1.05 V时基本达到极限电流区域,故选择1.05 V作为检测OA的工作电位。
2.4.2 线性范围与检出限在0~2 100 s范围内,分别考察了Pt NPs/GCE和Pt/GCE在连续加入不同浓度OA时的安培曲线(每个浓度平行加入3次,1.0 mmol/L的浓度平行加入5次)。在Pt NPs/GCE(图6曲线1)上,随OA浓度的增大,电流以台阶状稳态增大,电极对OA的响应时间小于5 s。电流与浓度在2.0×10-7~4.9×10-4mol/L和4.9×10-4~6.3×10-3mol/L范围内呈线性关系(插图A-1、B-1),线性方程分别为:I(μA)=0.003+0.083cOA(μmol/L)和I(μA)=60.24+0.061cOA(μmol/L),相关系数(r2)分别 为0.999 1和0.998 2,检 出 限(3sb)为2.7×10-8mol/L,灵敏度为83 μA/(mmol·L-1)。在Pt/GCE上(图6曲线2),OA也表现出快速的电流响应,且电流与浓度在1.1×10-6~2.4×10-4mol/L和2.4×10-4~6.3×10-3mol/L范围内呈线性关系(插图A-2、B-2),线性方程分别为:I(μA)=-0.012+0.039cOA(μmol/L)和I(μA)=1.790+0.029cOA(μmol/L),相关系数(r2)分别为0.991 8和0.999 3,检出限(3sb)为5.1×10-7mol/L,灵敏度为39 μA/(mmol·L-1)。对比显示,Pt NPs/GCE比Pt/GCE具有更好的分析性能。这是由于电化学去合金法制备的Pt纳米粒子具有更小的粒径和更大的活性面积。另一方面,相比于文献报道的基于Pt、Pd纳米材料的修饰电极(见表1),Pt NPs/GCE电极具有线性范围宽、灵敏度高及制备简单等特点。

图6 Pt NPs/GCE(曲线1)和Pt/GCE(曲线2)电极对OA的安培响应曲线Fig.6 Hydrodynamic amperometric responses of Pt NPs/GCE(curve 1)and Pt/GCE(curve 2)for OA concentration of OA(a-l):0.20,0.50,1.0,2.0,5.0,10.0,20.0,50.0,100,200,500,1 000 μmol/L,working potential:1.05 V;insert:the corresponding calibration plots for Pt NPs/GCE(A-1)and Pt/GCE(B-1)

表1 不同修饰电极测定草酸的分析性能比较Table 1 Comparison of the analytical performance of different modified electrodes for the determination of OA
2.4.3 干扰实验、稳定性与重现性研究了尿液中常见共存物质的干扰情况。结果表明,30.0 μmol/L OA条件下,1倍维生素C(Vc)、2倍胱氨酸、5倍柠檬酸、16倍肌酐、50倍氨水、100倍葡萄糖、500倍尿素的误差不超过±5%;100倍Zn2+、Mg2+、Ca2+,200倍NO-3、CO2-3、SO2-4、PO3-4,300倍K+、Na+、Cl-的误差不超过±5%。同样浓度尿酸的误差约为13%,但这种干扰可通过生成CaC2O4沉淀去除。表明该电极具有良好的选择性,可用于尿液中OA含量的测定。
采用同一支电极,重复5次测定30.0 μmol/L OA,电流响应的相对标准偏差(RSD)为3.3%,采用相同方法制备5支修饰电极测定30.0 μmol/L OA,电流响应的RSD为7.2%。电极干态保存2周后,测定同一浓度OA,电流响应几乎不变,上述结果表明该电极具有较好的稳定性与重现性。
2.5 实际样品的测定
以Pt NPs/GCE为工作电极测定尿液中OA的含量,并进行加标回收实验。样品制备参考文献[30]:采集新鲜尿液5.00 mL,以0.10 mol/L HClO4调至pH 5.0,准确移取2.00 mL该尿液于50 mL离心管中,依次加入0.40 mL 0.50 mol/L的CaCl2,8 mL无水乙醇,混匀后静置12 h,以4 000转离心15 min,弃去上清液,再向沉淀中加入20 mL 0.020 mmol/L乙酸-CaC2O4平衡液,混匀后离心15 min,弃上清液,加入2.00 mL水,混匀后离心15 min,弃上清液,沥干水分。最后加入0.50 mL 1.0 mol/L的HClO4溶解沉淀,制得测试液。按实验方法,向10.00 mL不断搅拌的支持电解质溶液中分别加入0.030 mL测试液和不同浓度的OA标准溶液,记录I~t曲线,测量电流,用标准曲线法计算OA的浓度,并计算加标回收率,结果如表2所示。经换算,两份尿液中的OA浓度分别为305 μmol/L和114 μmol/L,加标回收率分别为94.6%~102%和97.7%~101%,相对标准偏差分别为3.2%~4.7%和2.2%~3.8%。

表2 尿液中OA的测定结果及加标回收率(n=3)Table 2 Results of determination of OA in urine and its recoveries(n=3)
3 结论
本文以Bi为活泼金属,采用电化学合金-去合金法制备了一种多孔结构Pt纳米粒子修饰电极。该方法具有制备程序简单、产物的形貌和组成易控且可在常温下进行的优点。所制备电极对草酸的电氧化过程表现出高的催化活性。在此基础上,采用安培法定量测定人尿液中的OA含量。与同类电极相比,该Pt纳米粒子修饰电极具有灵敏度高和线性范围宽等特点。
