MTO 废弃催化剂资源化利用制备A 型分子筛
2023-01-04齐升东孙晓雪王德举
齐升东,孙晓雪,王德举,刘 丽
中国石油化工股份有限公司上海石油化工研究院,绿色化工与工业催化国家重点实验室,上海 201208
甲醇制低碳烯烃(MTO)是替代石油路线的新工艺[1-3]。MTO 技术采用流化床反应再生工艺,由于流化床反应再生技术的固有特性以及流化过程影响因素的多样性,固体催化剂在反应再生流动过程中剧烈撞击和摩擦,使催化剂破碎甚至粉化,催化剂细颗粒以及粉化颗粒被连续带出而废弃。
MTO 催化剂是典型的磷铝分子筛催化剂,因催化剂本身含磷且废弃催化剂微粉吸附较多的油类等污染物形成危险废物[4]。MTO 催化剂所含磷作为一种几乎不可再生、不可替代的国家战略性稀缺资源,又是引起水体富营养化、导致藻类繁殖的元素[5-6],如果不加以处理不仅对环境造成危害,而且造成资源浪费。MTO 废弃催化剂除含磷外,还含有较多的硅铝元素。因此,对MTO 废弃催化剂进行有效的回收利用,不仅符合环保要求,还可兼顾经济效益和社会效益。目前,废弃催化剂的资源化利用已形成产业,主要回收方法大致分为干法和湿法,也有企业采用干湿法结合或者简单处理后回用[7-8]。对废弃催化剂破碎、打浆再成型重新制备催化剂是资源化利用较好的途径,但难以兼顾催化剂活性和颗粒强度[9]。
开发MTO 废弃催化剂的资源化利用技术,变废为宝,化害为益,符合国家“减量化、再利用、资源化”的循环经济发展要求。本工作以硝酸为浸出剂对MTO 废弃催化剂进行浸出处理并控制剩余固渣硅铝比(物质的量之比),通过优化反应条件,采用碱熔活化-水热晶化转化硅铝固渣制备A 型分子筛。
1 实验部分
1.1 硝酸浸出处理
将MTO 废弃催化剂加入三口圆底烧瓶内,加入硝酸溶液,在实验温度下搅拌反应1 h。反应结束后,将反应产物进行抽滤,并用少量去离子水洗涤,浸出后固渣干燥备用。测定固渣中P,Al 和Si的含量,计算各元素的浸出率,计算公式如式(1)所示。

式中:x为浸出率,%;m1为废弃催化剂初始质量,g;m2为干燥固渣质量,g;ai为i元素在废催化剂中的质量分数,%;bi为i元素在浸出后固渣中的质量分数,%;i为P,Al 或Si。
1.2 A 型分子筛制备
将1.1 中干燥固渣与氢氧化钠按一定比例充分混合,在450 ℃下焙烧活化3 h,将活化后混合物料加入适量蒸馏水,进行陈化、晶化,产物经过滤、洗涤得到分子筛样品。
1.3 分析方法
采用德国布鲁克公司S4 PIONEER 型X 射线荧光光谱(XRF)分析样品的化学成分;采用Philips公司XL30E 型扫描电子显微镜(SEM)进行分子筛形貌的表征分析;采用日本Rigaku 公司D/max-1400型X 射线多晶粉末衍射(XRD)分析样品晶相结构,以Cu-Kα为放射源,在管电压为40 kV,管电流为40 mA 下扫描;采用美国Varian 公司725 ES 型电感耦合等离子体发射光谱仪(ICP-AES)进行固体物料中P,Al 和Si 元素含量分析。
1.4 静态水吸附容量测试
根据《分子筛静态水吸附测定方法》(GB/T 6287-2021)测定分子筛的静态水吸附容量。
2 结果与讨论
2.1 固渣硅铝比控制
以1 mol/L 硝酸溶液为浸出剂,在90 ℃下,采用不同液固比进行浸出研究,P 的浸出率及硅铝比变化如图1 所示。由图可知,随着液固比的增大,P 的浸出率与固渣硅铝比逐渐增大。当液固比由5升高至10 时,P 浸出率大幅度增大,继续增大液固比,P 浸出率增长缓慢。由结果可见液固比为5 的条件下,并不能保证硝酸与废催化剂的充分作用,P 的浸出率较低。在液固比为20 的条件下,P 的浸出率达到92%,同时浸出后固渣的硅铝比约为1,满足直接制备A 型分子筛的基本条件,但此条件下,浸出剂用量较大,相应滤液中P 元素的浓度较低,不利于后续P 资源的回收再利用。因此,综合各方面因素,后续研究采用液固比为10 进行优化。

图1 液固比对磷浸出的影响Fig.1 Effect of liquid-solid ratio on phosphorus leaching
在液固比为10,温度为90 ℃的条件下,考察硝酸浓度对P 浸出的影响,结果见图2。由图2 可知,随着硝酸浓度的增加,P 的浸出率和硅铝比均增加,当硝酸浓度为2 mol/L 时能够实现固渣的硅铝比为1,此时P 的浸出率达到93.73%。
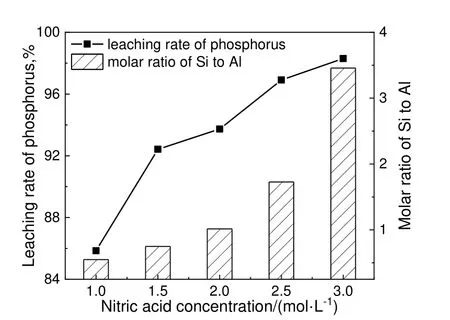
图2 硝酸浓度对磷浸出的影响Fig.2 Effect of nitric acid concentration on phosphorus leaching
在硝酸浓度为2 mol/L,液固比为10 的条件下,考察浸出温度对浸出效果的影响,结果见图3。从图3 可以看出,浸出温度的降低并未引起P 浸出率的大幅度改变,但固渣的硅铝比随浸出温度的降低而呈现降低的趋势。这表明在该条件下MTO 废弃催化剂中P 的浸出较为容易,但铝元素与催化剂中磷或硅形成较为稳固的结构浸出较为困难,且降低浸出温度减弱了传质速率,使铝的浸出效果降低,导致固渣的硅铝比降低。
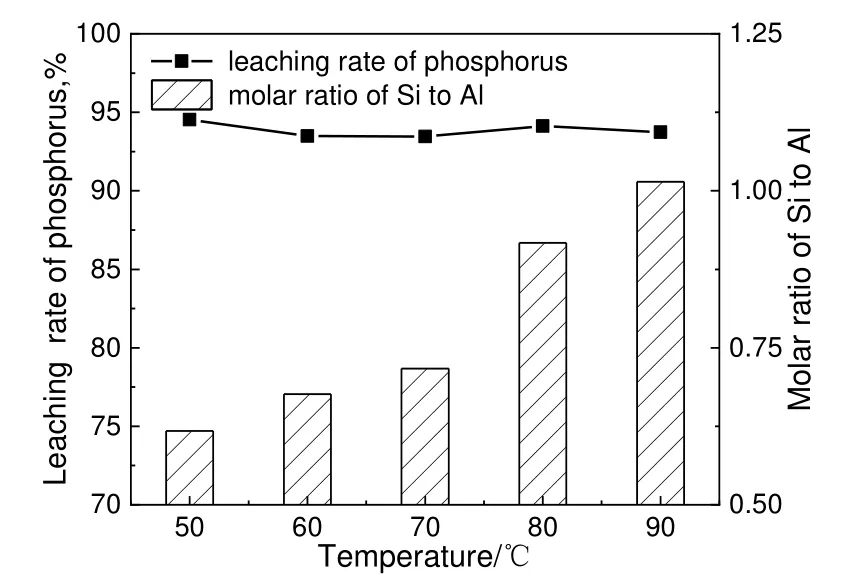
图3 浸出温度对磷浸出的影响Fig.3 Effect of temperature on phosphorus leaching
综上所述,以2 mol/L 的硝酸溶液为浸出液,在液固比为10,浸出温度为90 ℃的条件下与废弃催化剂作用,能够保证相对较高的P 元素浸出率并得到硅铝比约为1 的固渣,固渣可直接作为制备A 型分子筛的硅铝原料,后续分子筛的制备均采用此条件下的浸出固渣为硅铝源。
2.2 A 型分子筛制备
2.2.1 氢氧化钠用量的影响
A 型分子筛的重要用途之一是作为吸水剂使用,静态水吸附容量是商品A 型分子筛吸水剂的重要指标,所以后续研究中以吸水性能作为重要指标判断制备A 型分子筛样品的效果。
以浸出固渣为硅铝源,分别采用碱熔-水热和直接水热两种方式处理,在陈化时间为24 h,晶化温度为80 ℃,晶化时间为24 h 的条件下,考察不同NaOH 加入量(质量分数)对制备A 型分子筛吸水性能的影响,结果如图4 所示。由图可知,两种晶化方法制备的分子筛静态水吸附容量均随着氢氧化钠用量的增大先升高后降低,碱熔-水热法在碱用量为固渣量的60%时制备的分子筛静态水吸附容量较好,达到24.11%,直接水热法在碱用量为70%时较好,静态水吸附容量为19.12%。碱可促进固渣中硅铝物种向液相SiO32-和AlO2-转化,加快了液相凝胶向沸石晶体的转化速率[10]。分子筛样品静态水吸附容量达到最大时,水热法用碱量大于碱熔-水热法,分子筛吸水性能却低于碱熔-水热法,这是由于废弃催化剂中含有以石英等形式存在的SiO2,常规水热法并不能将硅铝源全部活化为硅铝酸盐,而碱熔处理可使石英等惰性物质晶体结构破坏,转变为可溶性的硅铝酸盐,进一步水热转化为A 型沸石,从而降低了最终产品中石英等惰性物质的含量,提高了A 型沸石的晶化程度[11]。因此,后续固定氢氧化钠添加量为60%来考察其他反应条件对碱熔-水热法制备A 型分子筛的影响。
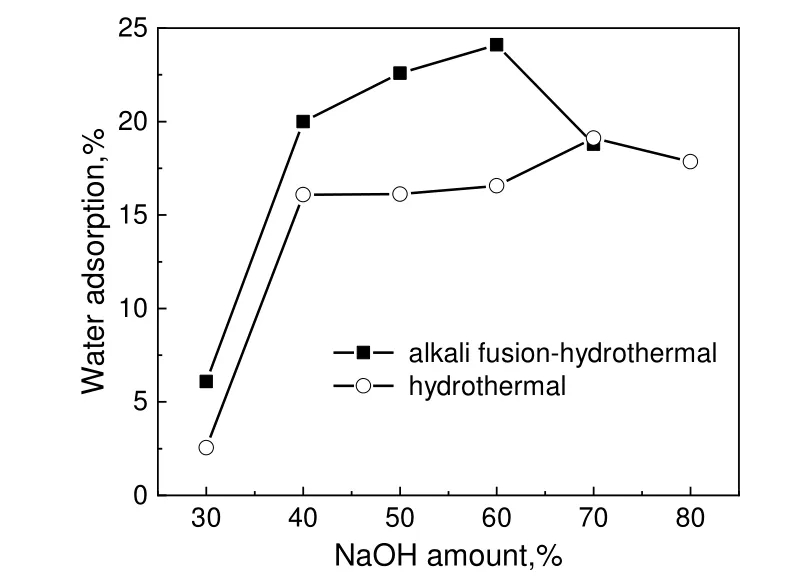
图4 氢氧化钠用量对分子筛吸水性能的影响Fig.4 Effect of NaOH amount on water adsorption of zeolites
2.2.2 水渣比的影响
水热晶化过程中H2O 使反应体系中的各种离子发生羟基化作用或水合作用形成羟基化离子或水合离子,加速反应组分的混合及移动,从而促使晶化反应的进行[12]。因此,晶化过程中水的用量对分子筛的制备有重要的影响。采用不同的水用量即在不同水渣比下制备分子筛,其XRD 图谱以及吸水性能结果分别见图5 和图6。由图5 可知,制备的分子筛均呈现典型的A 型分子筛衍射峰,水渣比为5 时,结晶度最高。由图6 可知,随着水渣比的增加,吸水率先增大后降低,在水渣比为5 时吸水率达到最大值。这是由于过小的水渣比会导致体系碱度过高,容易生成杂晶,导致吸水率较低[13];而水渣比过高时,体系碱度降低,制备的分子筛结晶度下降,静态水吸附容量降低。
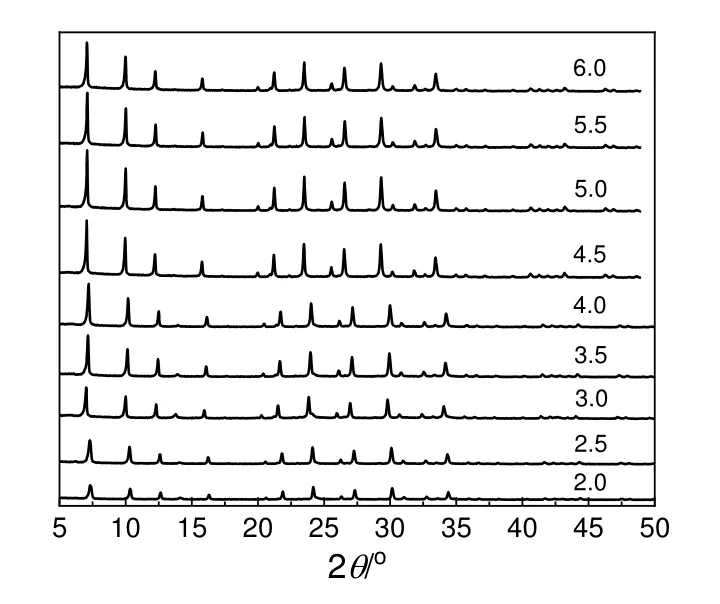
图5 不同水渣比下分子筛的XRD 图谱Fig.5 XRD patterns of zeolite with different water-slag ratio

图6 水渣比对分子筛吸水性能的影响Fig.6 Effects of water-slag ratio on water adsorption of zeolites
2.2.3 晶化温度及时间的影响
从原料的均匀混合到升温晶化前的静止过程称为陈化,这个过程影响着硅铝物质的溶解,进一步凝胶的组成和结构发生变化,这个过程还包含缓慢的分子筛成核过程。陈化时间过短,硅铝活性物质无法在碱性环境中完全溶解而形成凝胶[14];陈化时间过长,易生成杂晶,导致分子筛晶相不纯,从而影响分子筛的性能。通过实验确定较优的陈化时间并在后续实验中先陈化处理24 h 再升温晶化。图7是不同晶化温度处理24 h 制备分子筛的吸水性能曲线。由图7 可知,晶化温度较低时晶化不充分,制备的分子筛样品吸水率较小;晶化温度为80 ℃制备的分子筛样品静态水吸附容量达到24.11%;继续升高晶化温度,导致杂晶的产生,静态水吸附容量迅速下降。可见,晶化温度过低或过高均会降低产品的吸水率。这主要是因为晶化温度过低导致分子筛晶化不完全,而晶化温度过高导致杂晶生成[15]。
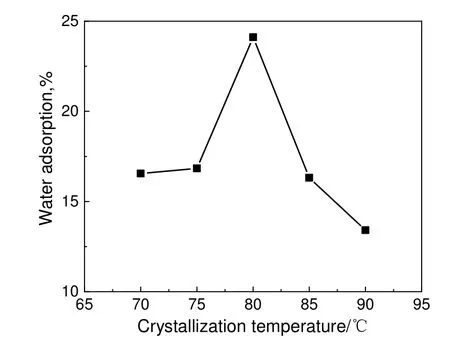
图7 晶化温度对分子筛吸水性能的影响Fig.7 Effects of crystallization temperature on water adsorption of zeolites
晶化时间也是影响晶体生长和转化的重要因素,通常情况下分子筛晶体随着晶化时间的延长而不断生长[16]。图8 为A 型分子筛吸水性能与晶化时间的关系。由图8 可知,晶化时间从12 h 延长到24 h,制备的分子筛静态水吸附容量快速升高。这是因为经过成核诱导期后,12~24 h 的晶化时间内A 型分子筛在生长期快速生成并长大,导致产品的静态水吸附容量迅速提高,图9 中样品XRD 峰强度从12 h 至24 h 迅速升高的变化也印证了A 型分子筛的快速生成这一现象[17]。继续延长晶化时间,产品的静态水吸附容量和A 型分子筛的XRD 峰强度基本不变,这说明晶化时间24 h 后,物料转化基本完成已获得结晶完全的A 型分子筛,此时体系中的晶体生长速度和溶解速度相近即达到动态平衡。晶化时间大于24 h 后,晶体大小会发生变化,但对样品结晶度和静态水吸附容量影响较小[11]。继续延长晶化时间至36 h 及以上时,XRD 谱图2θ为14.091°处出现了微弱的方钠石特征衍射峰,这表明少量A 型分子筛进一步转化生成方钠石。因为方钠石是一种致密结构的分子筛,其孔径更小不能吸附水分子,所以导致分子筛吸水剂性能略有降低[18]。实验结果表明,浸出固渣制备A 型分子筛较优的晶化条件为室温陈化24 h,于80 ℃晶化24 h。优化条件下制备的A 型分子筛静态水吸附容量大于24%,达到《4A 分子筛》(HG/T 2524-2010)中一等品的吸水率标准。
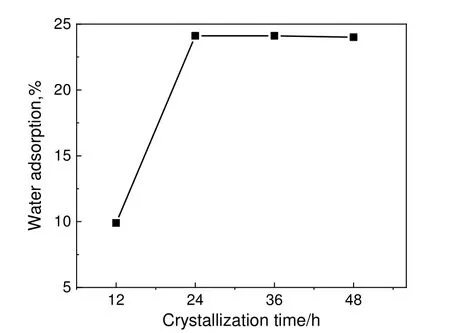
图8 晶化时间对分子筛吸水性能的影响Fig.8 Effects of crystallization time on water adsorption of zeolites

图9 不同晶化时间下分子筛XRD 图谱Fig.9 XRD patterns of zeolite with different crystallization times
2.3 A 型分子筛表征分析
图10 为酸浸-碱熔-晶化三个不同阶段固体样品的表面形貌。图10(a)是经硝酸处理后的固渣,由于硝酸作用,破坏了MTO 废弃催化剂的结构与形貌,部分位置形成较大孔隙,整体形貌较为疏松,以不规则片状居多;当以氢氧化钠碱熔后,固渣中剩余的无定型硅铝与碱发生熔融作用,形成图10(b)中较为致密的沟壑状形貌;制备的样品中形成了图10(c)中典型A 型分子筛的立方晶体结构,晶粒直径为1.5~2.0 μm,立方晶粒呈堆砌状。
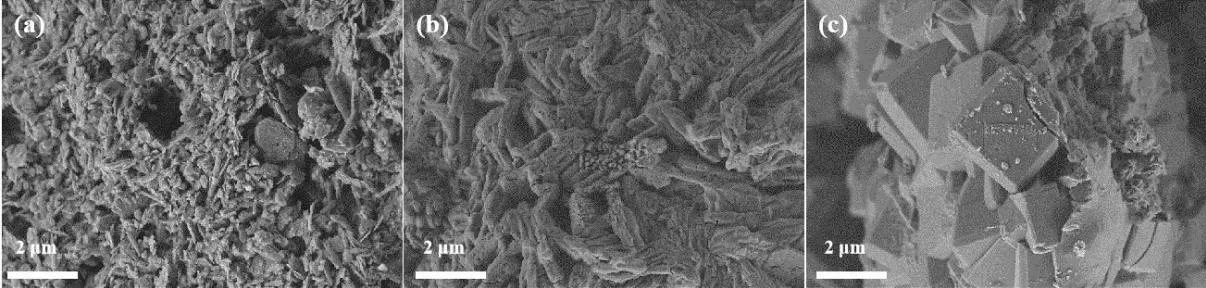
图10 不同阶段样品SEM 照片Fig.10 SEM images of samples at different stages
3 结 论
a)以2 mol/L 的硝酸溶液,在浸出液固比为10,浸出温度为90 ℃条件下与废弃催化剂反应,P的浸出率在90%以上,得到硅铝比约为1 的固渣,可直接作为制备A 型分子筛的硅铝原料。
b)向固渣中加入60%的碱,于450 ℃下活化3 h,在水渣比为5 的条件下陈化24 h,80 ℃晶化24 h 制备的A 型分子筛静态水吸附容量大于24%,达到一等品4A 分子筛活化粉的性能标准。
