色胺酮衍生物无金属催化的高效合成及抗菌活性研究
2022-12-30刘万聪王亚鹏辛子杰徐鹏帅代海渝邓力超王启卫1
陈 林, 刘万聪, 王亚鹏, 辛子杰, 徐鹏帅,代海渝, 邓力超, 王启卫1,,4*, 张 翔*
(1. 中国科学院 成都有机化学研究所,四川 成都 610041;2. 成都大学 药学院(川抗所) 抗生素研究与再评价四川省重点实验室,四川 成都 610106;3. 中国科学院大学,北京 100049; 4. 西华大学 理学院,四川 成都 610039)
色胺酮又名板蓝根二酮B,最早是在1971年由Schindle等从过量L-色氨酸存在下的解脂假丝酵母(Candida lipolytica)中分离出来的喹唑啉酮类生物碱[1-2]。色胺酮主要存在于中药青黛、大青叶中,也是药用植物马蓝、蓼蓝和菘蓝等产蓝植物的主要活性成分之一[3-5]。色胺酮独特的杂环骨架赋予了良好的药理活性,如抗炎[6]、抗菌[7]、抗肿瘤[8-9]和调节免疫功能[10]等。色胺酮对疟疾、利什曼原虫和肺结核病菌等具有突出的药物活性[11-12]。同时,色胺酮的吲哚和喹唑啉酮母核等亚结构也是许多天然生物碱和药物的重要组成部分[13-14](Scheme 1a)。因此,开发简单、有效和易于放大生产的合成方法,制备结构多样的色胺酮衍生物,不仅具有重要的理论意义,还具有较好的实际价值。
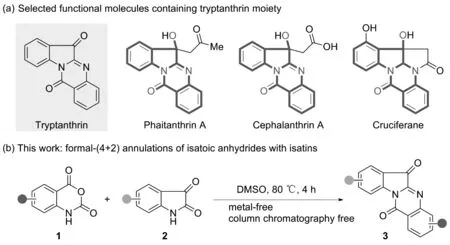
Scheme 1
基于色胺酮良好的药物活性,近年来,其相关的合成方法研究取得了一定进展。以靛红作为单一的起始原料,通过不同方法引发的自缩合反应可以得到色胺酮[15-18]。在不同的催化剂体系作用下,取代吲哚同样能够转化为色胺酮衍生物[19-22]。此外,邻氨基苯甲酸与靛红或邻氨基苯乙酮与靛红酸酐的环化反应也可被用于色胺酮衍生物的合成[23-25]。然而,这些方法大多使用强毒性氧化剂、依赖过渡金属作为催化剂且需要高温等较为苛刻的反应条件。尽管在一些反应条件下,通过靛红或吲哚与靛红酸酐的缩合反应,也可以得到色胺酮的衍生物[12,26-32],但其柱层析的后处理过程增加了这些方法在放大生产中的成本,限制了其应用范围。
基于本课题组前期对于色胺酮修饰与改造的研究[33],本文采用取代靛红酸酐1作为起始原料,DMSO作为反应溶剂,在DABCO的作用下,以80 ℃作为反应温度,与取代靛红2经形式上的(4+2)环化反应以良好到优秀的收率合成了一系列色胺酮衍生物。在后处理过程中,无需柱层析,经简单便捷的过滤、水洗和干燥等操作即可得到目标产物,该方法在减小对环境影响的同时提升了方法的实用性与合成效率(Scheme 1)。所得化合物均通过1H NMR,13C NMR和HR-MS(ESI-TOF)进行表征。最后,探究了DABCO的作用机理,并通过最小抑菌浓度(MIC)试验对其良好的体外抗菌活性进行了初步的评价。
1 实验部分
1.1 仪器与试剂
WRX-X-4A型熔点仪;JEOL-600 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Waters SYNAPT G2型高分辨质谱仪。
所用试剂均为分析纯。
1.2 色胺酮衍生物3a~3l的合成(以3a为例)
在反应试管中依次加入靛红酸酐1a(0.12 mmol, 19.60 mg)、靛红2a(0.10 mmol, 14.70 mg)及DABCO(0.02 mmol, 2.20 mg)。随后,加入1.00 mL二甲基亚砜,并于80 ℃条件下反应4 h(TLC监测2a消耗完毕),过滤,滤饼用水洗涤,真空干燥得3a。用类似的方法合成3b~3l。
吲哚并[2,1-b]喹唑啉-6,12-二酮(3a):黄色固体,收率92%, m.p.264~267 ℃;1H NMR(600 MHz, CDCl3)δ: 8.61(d,J=7.2 Hz, 1H), 8.42(dd,J=8.4 Hz, 1.2 Hz, 1H), 8.02(d,J=8.4 Hz, 1H), 7.91(d,J=7.8 Hz, 1H), 7.84~7.87(m, 1H), 7.77~7.80(m, 1H), 7.67(t,J=7.8 Hz, 1H), 7.43(t,J=7.2 Hz, 1H);13C NMR(151 MHz, CDCl3)δ: 182.5, 158.0, 146.6, 146.3, 144.3, 138.3, 135.1, 130.7, 130.2, 127.5, 127.2, 125.4, 123.7, 121.9, 117.9; HR-MS(ESI)m/z: calculated for C15H8N2O2{[M+Na]+}271.0478, found 271.0480。
2-氯-吲哚并[2,1-b]喹唑啉-6,12-二酮(3b):黄色固体,收率90%, m.p.>300 ℃;1H NMR(600 MHz, CDCl3)δ: 8.63(d,J=8.4 Hz, 1H), 8.41(s, 1H), 7.98(d,J=7.8 Hz, 1H), 7.94(d,J=7.2 Hz, 1H), 7.79~7.83(m, 2H), 7.46(t,J=7.8 Hz, 1H);13C NMR(151 MHz, CDCl3)δ: 182.5, 157.2, 146.1, 145.2, 138.5, 136.7, 135.6, 132.2, 127.6, 127.3, 125.6, 125.0, 122.0, 118.1; HR-MS(ESI)m/z: calculated for C15H7ClN2O2{[M+H]+}283.0269, found 283.0266。
2-溴-吲哚并[2,1-b]喹唑啉-6,12-二酮(3c):黄色固体,收率92%, m.p.>300 ℃;1H NMR(600 MHz, CDCl3)δ: 8.62(d,J=8.4 Hz, 1H), 8.58(s, 1H), 7.90~7.96(m, 3H), 7.82(t,J=7.2 Hz, 1H), 7.46(t,J=6.6 Hz, 1H);13C NMR(151 MHz, CDCl3)δ: 182.2, 156.9, 146.0, 145.2, 144.4, 138.4, 138.4, 132.1, 130.3, 127.5, 125.5, 125.1, 124.7, 121.9, 118.1; HR-MS(ESI)m/z: calculated for C15H7BrN2O2{[M+H]+}326.9764, found 326.9756。
2-甲基-吲哚并[2,1-b]喹唑啉-6,12-二酮(3d):黄色固体,收率83%, m.p.254~256 ℃;1H NMR(600 MHz, CDCl3)δ: 8.61(d,J=8.4 Hz, 1H), 8.21(s, 1H), 7.91(m, 2H), 7.76~7.79(m, 1H), 7.65(dd,J=2.4 Hz, 8.4 Hz, 1H), 7.42(t,J=7.2 Hz, 1H), 2.55(s, 3H);13C NMR(151 MHz, CDCl3)δ: 182.6, 158.1, 146.2, 144.5, 143.7, 141.3, 138.1, 136.4, 130.5, 127.3, 127.1, 125.3, 123.4, 122.0, 117.9, 21.7; HR-MS(ESI)m/z: calculated for C16H10N2O2{[M+Na]+}285.0634, found 285.0638。
3-氯-吲哚并[2,1-b]喹唑啉-6,12-二酮(3e):黄色固体,收率93%, m.p.272~276 ℃;1H NMR(600 MHz, CDCl3)δ: 8.61(d,J=8.4 Hz, 1H), 8.37(d,J=8.4 Hz, 1H), 8.01(s, 1H), 7.93(d,J=6.6 Hz, 1H), 7.79~7.82(m, 1H), 7.63(dd,J=2.4 Hz, 8.4 Hz, 1H), 7.45(t,J=7.8 Hz, 1H);13C NMR(151 MHz, CDCl3)δ: 182.2, 157.5, 147.6, 146.2, 145.2, 141.5, 138.5, 130.7, 130.1, 128.8, 127.4, 125.6, 122.2, 121.7, 118.0; HR-MS(ESI)m/z: calculated for C15H7ClN2O2{[M+H]+}283.0269, found 283.0268。
8-氟-吲哚并[2,1-b]喹唑啉-6,12-二酮(3f):黄色固体,收率88%, m.p.272~274 ℃;1H NMR(600 MHz, CDCl3)δ: 8.64(dd,J=4.2 Hz, 9.0 Hz, 1H), 8.43(d,J=8.4 Hz, 1H), 8.03(d,J=7.2 Hz, 1H), 7.85~7.88(m, 1H), 7.68~7.71(m, 1H), 7.58(dd,J=2.4 Hz, 6.6 Hz, 1H), 7.49(t,J=8.4 Hz, 1H);13C NMR(151 MHz, CDCl3)δ: 181.7, 161.0(J=251.6 Hz), 157.9, 146.5, 144.3, 142.4, 135.2, 130.8, 130.5, 127.5, 124.8(J=24.6 Hz), 123.7, 123.3, 119.6(J=7.2 Hz), 112.1(J=24.5 Hz);19F NMR(565 MHz, CDCl3)δ: -112.3; HR-MS(ESI)m/z: calculated for C15H7FN2O2{[M+Na]+}289.0384, found 289.0375。
8-氯-吲哚并[2,1-b]喹唑啉-6,12-二酮(3g):黄色固体,收率94%, m.p.291~294 ℃;1H NMR(600 MHz, DMSO-d6)δ: 8.43(d,J=9.0 Hz, 1H), 8.29(d,J=7.8 Hz, 1H), 7.93(m,J=2.8 Hz, 3H), 7.88(dd,J=2.4 Hz, 8.4 Hz, 1H), 7.70~7.73(m, 1H);13C NMR(151 MHz, DMSO-d6)δ: 181.9, 158.2, 146.9, 145.5, 145.0, 137.4, 135.9, 131.7, 130.6, 130.5, 127.5, 124.8, 124.6, 123.6, 119.1; HR-MS(ESI)m/z: calculated for C15H7ClN2O2{[M+H]+}283.0269, found 283.0270。
8-溴-吲哚并[2,1-b]喹唑啉-6,12-二酮(3h):黄色固体,收率86%, m.p.282~285 ℃;1H NMR(600 MHz, CDCl3)δ: 8.53(d,J=7.8 Hz, 1H), 8.43(d,J=8.3 Hz, 1H), 8.02~8.04(m, 2H), 7.88(m, 2H), 7.68~7.71(m, 1H);13C NMR(151 MHz, CDCl3)δ: 181.3, 157.9, 146.5, 144.9, 143.7, 140.6, 135.3, 130.9, 130.6, 128.2, 127.6, 123.6, 123.3, 120.7, 119.5; HR-MS(ESI)m/z: calculated for C15H7BrN2O2{[M+H]+}326.9764, found 326.9759。
8-甲基-吲哚并[2,1-b]喹唑啉-6,12-二酮(3i):黄色固体,收率81%, m.p.275~278 ℃;1H NMR(600 MHz, CDCl3)δ: 8.47(d,J=8.4 Hz, 1H), 8.42(d,J=6.6 Hz, 1H), 8.01(d,J=8.4 Hz, 1H), 7.81~7.84(m, 1H), 7.69(s, 1H), 7.65(t,J=7.2 Hz, 1H), 7.56(d,J=8.4 Hz, 1H), 2.45(s, 3H);13C NMR(151 MHz, CDCl3)δ: 182.6, 157.9, 146.8, 144.7, 144.4, 138.8, 137.4, 134.9, 130.7, 130.0, 127.5, 125.4, 123.9, 122.2, 117.7, 21.0; HR-MS(ESI)m/z: calculated for C16H10N2O2{[M+Na]+}285.0634, found 285.0641。
8-甲氧基-吲哚并[2,1-b]喹唑啉-6,12-二酮(3j):黄色固体,收率92%, m.p.265~269 ℃;1H NMR(600 MHz, CDCl3)δ: 8.50(d,J=9.0 Hz, 1H), 8.41(d,J=7.2 Hz, 1H), 8.01(d,J=7.2 Hz, 1H), 7.82(t,J=7.8 Hz, 1H), 7.65(t,J=7.8 Hz, 1H), 7.37(d,J=2.4 Hz, 1H), 7.29(dd,J=3.0 Hz, 9.0 Hz, 1H), 3.90(s, 3H);13C NMR(151 MHz, CDCl3)δ: 182.5, 158.9, 157.7, 146.8, 144.8, 140.5, 134.8, 130.7, 130.1, 127.4, 124.9, 124.0, 123.1, 119.2, 108.6, 56.0; HR-MS(ESI)m/z: calculated for C16H10N2O3{[M+Na]+}301.0584, found 301.0586。
9-氯-吲哚并[2,1-b]喹唑啉-6,12-二酮(3k):黄色固体,收率90%, m.p.261~265 ℃;1H NMR(600 MHz, CDCl3)δ: 8.70(d,J=2.4 Hz, 1H), 8.45(dd,J=1.8 Hz, 8.4 Hz, 1H), 8.05(d,J=7.2 Hz, 1H), 7.85~7.90(m, 2H), 7.71(t,J=7.8 Hz, 1H), 7.42(dd,J=1.8, 7.2 Hz, 1H);13C NMR(151 MHz, CDCl3)δ: 181.1, 158.0, 146.8, 146.5, 144.7, 144.2, 135.4, 130.8, 130.5, 127.7, 127.6, 126.3, 123.4, 120.3, 118.6; HR-MS(ESI)m/z: calculated for C15H7ClN2O2{[M+H]+}283.0269, found 283.0267。
9-溴-吲哚并[2,1-b]喹唑啉-6,12-二酮(3l):黄色固体,收率85%, m.p.228~242 ℃;1H NMR(600 MHz, DMSO-d6)δ: 8.59(s, 1H), 8.28(d,J=7.8 Hz, 1H), 7.92(t,J=4.2 Hz, 2H), 7.80(d,J=8.4 Hz, 1H), 7.71~7.73(m, 1H), 7.67(dd,J=1.8 Hz, 8.4 Hz, 1H);13C NMR(151 MHz, DMSO-d6)δ: 182.0, 158.2, 147.0, 146.9, 145.5, 136.0, 131.3, 130.6, 130.5, 130.4, 127.6, 126.8, 123.5, 122.1, 120.2; HR-MS(ESI)m/z: calculated for C15H7BrN2O2{[M+H]+}326.9764, found 326.9764。
1.3 体外抗菌活性测试
采用美国临床和实验室标准协会(CLSI)抗菌药物敏感性试验操作规程(M02-A11, M07-A9和M11-A8)推荐的微量肉汤2倍稀释法测定了化合物3a~3l及对照药左氧氟沙星对耐甲氧西林金黄色葡萄球菌(MRSA)、敏感金黄色葡萄球菌(MSSA)、耐甲氧西林表面葡萄球菌(MRSE)和敏感表面葡萄球菌(MSSE)的最低抑菌浓度(MIC)。
2 结果与讨论
2.1 反应条件的优化
以3a的合成反应为模板反应,对反应条件进行了考察(表1)。首先,以DABCO为碱,在60 ℃条件下,对反应溶剂进行了筛选(Entry 1~8)。为了便于比较反应的情况,以柱层析分离收率为指标对反应进行考察,结果发现,反应结果与溶剂的极性呈现出一定的正相关性。尽管在使用乙腈、乙酸乙酯(EA)和二甲基亚砜(DMSO)时,反应均能以优秀的收率分离得到目标产物,但仅有当使用DMSO作为反应溶剂时,产物能够从反应体系中大量析出。基于此,DMSO有望作为一种可以简化后处理操作的反应溶剂。

表1 合成3a的反应条件优化Table 1 Optimization of the synthesis condition of 3a
考虑到反应过程中靛红酸酐需要脱除一分子的CO2,因此进一步升高反应温度至80 ℃以促进CO2释放,进而促进反应发生(Entry 10)。结果显示,升温不仅能够对反应收率起到一定程度的改善作用,也能进一步提升反应的效率。随后,在该温度下,进一步探索不同碱对反应的影响(Entry 11~13)。考虑到碱的溶解性问题,本文重点对有机碱进行了筛选。尽管在不同有机碱的催化作用下,反应均能够发生,但其结果与DABCO相比,均存在较为明显的差异。最后,本研究确定了反应的最优条件:以DMSO为溶剂,DABCO为碱,于80 ℃条件下进行反应(表1)。同时,本文进一步尝试不经柱层析,直接进行过滤、水洗和干燥的后处理操作,结果发现,反应收率并未受到显著影响(92%,表1, Entry 10)。
2.2 反应的底物扩展
在优化后的条件下,对反应底物的普适性进行了研究(Scheme 2)。首先以含有不同取代基的靛红酸酐为底物进行反应,均能以良好到优秀的收率(83%~93%)分离得到目标化合物3b~3e。之后进一步对取代靛红的底物兼容性进行考查,结果发现,反应同样能够顺利发生(81%~94%),并得到相应的目标产物(3f~3l)。特别地是,当以5-氯靛红作为反应底物时,该反应能够以94%收率过滤得到目标化合物,进一步论证了该方法的实用性和高效性。

Scheme 2
2.3 反应机理研究
在上述底物扩展的基础上,以DABCO在反应中的作用作为切入点,对反应的机理进行初步研究。根据已有文献报道[34],DABCO在反应中可以作为布朗斯特碱活化靛红,同时,本研究认为其还有可能作为路易斯碱活化靛红酸酐。因此,使用布朗斯特碱性较弱而路易斯碱性较强的三苯基膦作为催化剂,在标准条件下对反应进行探究,结果发现,反应仍然能够顺利进行,并以42%柱层析收率分离得到目标产物(Scheme 3)。结合近期文献报道的靛红酸酐的活化方式[35],该实验结果从侧面佐证了DABCO作为路易斯碱活化靛红酸酐的作用机理。

Scheme 3
据此,本文提出了上述环化反应可能的作用机理(Scheme 4):在反应过程中,DABCO不仅可以作为路易斯碱活化靛红酸酐生成两性离子中间体I,同时还能够作为布朗斯特碱拔除靛红的质子生成靛红负离子II;随后,中间体I和II中的氮负离子对彼此的羰基亲核进攻,质子化后形成中间体III;最终脱水形成色胺酮3a。

Scheme 4
2.4 体外抗菌活性
为进一步论证产物的有用性,对合成的色胺酮衍生物进行了体外抗菌活性评价。由表2可见,所有的化合物(3a~3l)对耐甲氧西林表面葡萄球菌(MRSE)、敏感表面葡萄球菌(MSSE)均能表现出较好的抑菌活性。基于4种致病菌的筛选结果,发现化合物3b与3g表现出一定的广谱抑菌活性,其中化合物3g的抑菌活性较好,化合物3g对4种致病菌的MIC值均小于4 μg·mL-1。
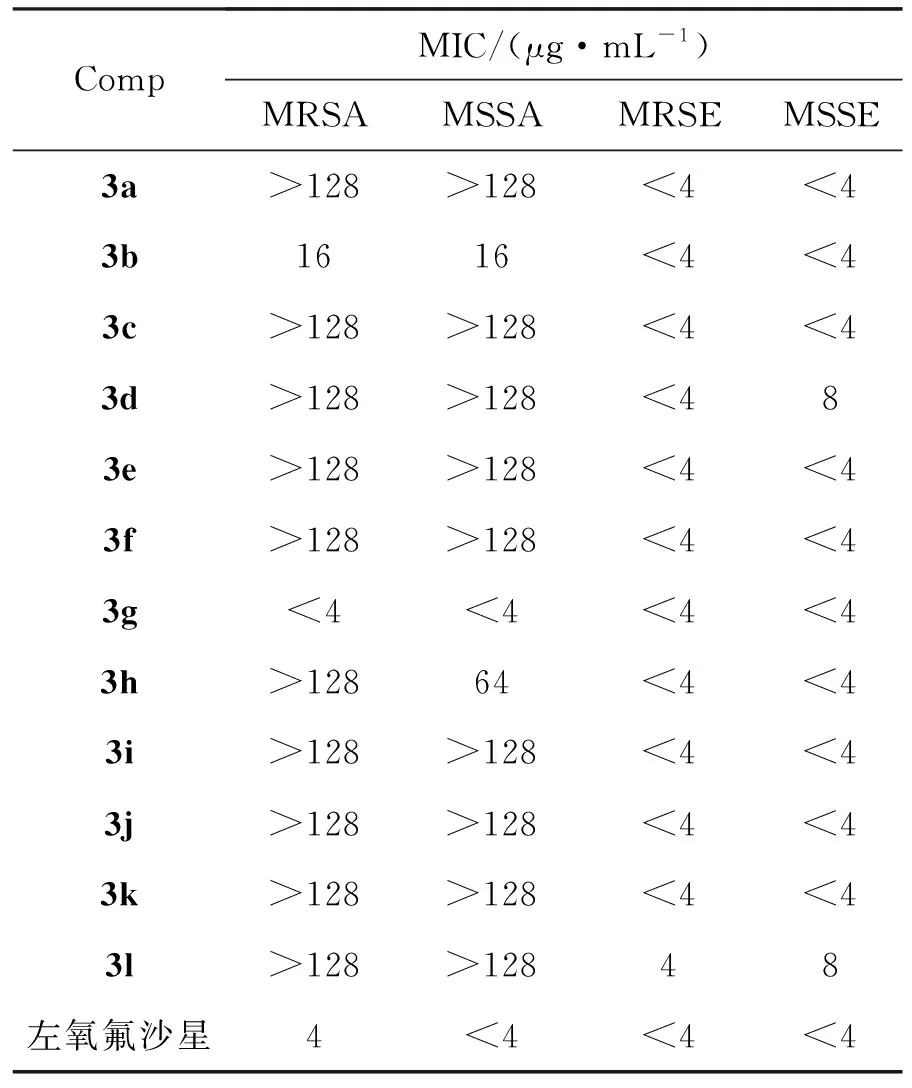
表2 3a~3l的体外抗菌活性Table 2 In vitro antimicrobial activities of 3a~3l
在DABCO的作用下,以80 ℃作为反应温度,采用取代靛红酸酐作为起始原料,DMSO作为反应溶剂,与取代靛红经形式上的(4+2)环化反应并以81%~94%收率合成了一系列色胺酮衍生物。在后处理过程中,无需柱层析,经简单便捷的过滤、水洗和干燥等操作即可得到目标产物。该方法进一步简化了合成途径、降低了合成成本,为色胺酮衍生物的高效制备提供了一种绿色、实用的工艺路线。在此基础上,探索了DABCO的作用机理,并通过最小抑菌浓度(MIC)试验初步揭示了化合物3g良好的体外抗菌活性(<4 μg·mL-1),而深入的构效分析研究仍在进行中。
