厚朴排气合剂主要药效成分在不同种属动物血浆中蛋白结合率的比较Δ
2022-12-29郭明鑫范文韬吴霞沈颖胡志强江苏大学附属宜兴医院药学部江苏宜兴400广东药科大学新药研发中心广州50006
郭明鑫,范文韬吴霞,沈颖胡志强(.江苏大学附属宜兴医院药学部,江苏 宜兴 400;.广东药科大学新药研发中心,广州 50006)
厚朴排气合剂由厚朴、枳实、木香、大黄组成,具有和中理气、行气消胀的功效,主要用于腹部非胃肠吻合术后早期肠麻痹的辅助治疗[1]。目前,该合剂可用于恢复患者术后胃肠道功能和治疗功能性便秘、肠麻痹等症[2]。有研究指出,厚朴排气合剂中厚朴酚含量最高,约占总成分含量的90%,和厚朴酚次之,同时还有少量的大黄素和大黄酚;其中,厚朴酚、和厚朴酚能促进肠道蠕动,是厚朴排气合剂的主要药效成分[3-4]。
血浆蛋白结合率会直接影响体内游离药物的浓度,从而影响药物的吸收、分布、代谢和消除,最终影响其药效学特征[5-6]。考虑到厚朴排气合剂单次给药剂量较大、相关不良反应较多[7],故有必要对其主要药效成分的血浆蛋白结合率进行测定,以有助于研究该合剂的体内代谢过程,促进临床合理用药。基于此,本研究拟采用超高效液相色谱-串联三重四极杆质谱(UPLC-MS/MS)技术结合平衡透析法测定厚朴排气合剂主要药效成分厚朴酚、和厚朴酚在牛、兔、大鼠血浆中的蛋白结合率并进行比较,旨在为阐明该合剂有效成分的血浆蛋白结合特性提供参考。
1 材料
1.1 主要仪器
本研究所用主要仪器包括API 5500型UPLC-MS/MS系统(美国AB Sciex公司)、BP210D型万分之一电子天平(德国Sartorius公司)、20HR型高速冷冻离心机(科大创新股份有限公司)、ZH-2型涡旋振荡仪(上海精胜科学仪器有限公司)、MG-2200型氮吹仪(上海爱郎仪器有限公司)等。
1.2 主要药品与试剂
厚朴排气合剂(批号20080601,每1 mL相当于饮片0.8 g)购自瑞阳制药股份有限公司;厚朴酚对照品(批号110729-201910,纯度99.0%)、和厚朴酚对照品(批号110730-201910,纯度99.8%)均购自中国食品药品检定研究院;牛血浆(批号J28S10S99034)、兔血浆(批号J10O10S99437)、磷酸盐 缓冲液(pH7.2~7.4,批号8121556)均购自上海源叶生物科技有限公司;大鼠血浆采自SPF级SD大鼠(雌雄各半);透析袋(截留分子量1 kDa)购自广州瑞舒生物科技有限公司;甲醇、乙腈、甲酸为质谱纯,其余试剂均为分析纯,水为纯净水。
2 方法与结果
2.1 溶液的制备
2.1.1 空白透析液 取磷酸盐缓冲液,加水稀释并定容至500 mL,作为空白透析液,于4 ℃下保存,备用。
2.1.2 含药透析液 分别取厚朴排气合剂适量,加至“2.1.1”项下空白透析液中,混匀,得质量浓度分别为4.5、9.0、13.5 μg/mL(按饮片质量计)的含药透析液。
2.1.3 混合工作液和质控样品 精密称定厚朴酚、和厚朴酚对照品适量,置于10 mL容量瓶中,用甲醇定容,配制成混合对照品储备液;精密量取上述混合对照品储备液1.00、0.80、0.64、0.36、0.28、0.10、0.03 mL,分别置于10 mL容量瓶中,用甲醇定容,混匀,配制成厚朴酚质量 浓 度 分 别 为 11.38、22.76、45.52、91.04、136.56、182.08、227.60 μg/mL,和厚朴酚质量浓度分别为3.90、7.80、15.60、31.20、46.80、62.40、78.00 μg/mL的系列混合工作液。精密量取混合对照品储备液适量,加入空白血浆,配制成厚朴酚质量浓度分别为1.14、2.28、9.10、18.21 μg/mL,和厚朴酚质量浓度分别为0.39、0.78、3.12、6.24 μg/mL的质控样品。上述溶液均于4 ℃下保存,备用。
2.2 样品预处理方法
2.2.1 透析内液 吸取平衡透析实验中的透析内液200 μL,置于1.5 mL EP管中,加入乙腈600 μL沉淀蛋白,涡旋振荡3 min后以12 000 r/min离心10 min。取上清液,以氮气流吹干,残渣用甲醇200 μL复溶,涡旋振荡3 min后以12 000 r/min离心10 min,取上清液进样测定。
2.2.2 透析外液 吸取平衡透析实验中的透析外液200 μL,置于1.5 mL EP管中,以12 000 r/min离心10 min,取上清液进样测定。
2.3 色谱与质谱条件
以ACQUITY UPLC BEH-C18(2.1 mm×50 mm,1.7 μm)为色谱柱,以乙腈(A)-0.1%甲酸溶液(B)为流动相进行梯度洗脱(0~3 min,65%A→75%A);流速为0.3 mL/min;柱温为40 ℃;进样量为2 μL。
采用电喷雾离子源(electrospray ionization,ESI),以选择反应监测(selective reaction monitoring,SRM)模式进行负离子扫描;离子传输管温度为325 ℃;气化室温度为350 ℃;电喷雾电压为3 kV;鞘气流速为50 Arb;辅助气流速为10 Arb;离子扫描范围为m/z50→1 500。用于定量分析的离子对分别为m/z264.4→247.1、244.9(厚朴酚,去簇电压分别为-116、-110 V,碰撞电压分别为-31、-34 V)和m/z264.4→246.9、223.1(和厚朴酚,去簇电压分别为-110、-100 V,碰撞电压分别为-28、-40 V)。
2.4 方法学考察
依照2020年版《中国药典》(四部)(以下简称“药典”)通则“生物样品定量分析方法验证指导原则”的相关要求进行方法学考察[8]。
2.4.1 专属性试验 分别取空白血浆、空白血浆+混合对照品溶液(厚朴酚、和厚朴酚的最终质量浓度分别为2.28、0.78 μg/mL)、平衡透析实验的透析内液样品(9.0 μg/mL含药透析液平衡透析24 h后所得),按照“2.2.1”项下方法处理后,再按照“2.3”项下条件进样测定,记录色谱图。结果显示,在上述条件下,厚朴酚、和厚朴酚色谱峰峰形良好,其保留时间分别约为1.3、1.0 min;血浆中内源性杂质不会对上述成分的定量测定产生干扰。结果见图1(因不同种属血浆样品的色谱图基本一致,故以空白牛血浆+混合对照品、牛血浆透析内液样品的色谱图作为代表)。

图1 厚朴酚、和厚朴酚专属性试验的典型SRM图
2.4.2 样本残留考察 本研究通过测定较高质量浓度质控样品(厚朴酚、和厚朴酚的质量浓度分别为18.21、6.24 μg/mL)来验证是否有残留。结果显示,测定高质量浓度质控样品后,空白血浆样品中的残留不超过定量下限的20%,符合药典相关要求[8]。
2.4.3 线性关系和定量下限考察 分别精密量取系列混合工作液10.00 μL,加至空白牛、兔、大鼠血浆90 μL中,得厚朴酚质量浓度分别为 1.14、2.28、4.56、9.12、13.65、18.24、22.76 μg/mL,和厚朴酚质量浓度分别为0.39、0.78、1.56、3.12、4.68、6.24、7.80 μg/mL的系列标准曲线样品,按照“2.2.1”项下方法处理后,再按照“2.3”项下条件进样测定,记录峰面积。以待测成分峰面积为纵坐标(y)、其质量浓度为横坐标(x)进行线性回归(权重系数为1/x2),分别拟合得厚朴酚、和厚朴酚的回归方程,并以线性范围下限作为定量下限。结果见表1。

表1 不同基质中厚朴酚、和厚朴酚定量分析的回归方程与线性范围
2.4.4 精密度与准确度试验 按照“2.1.3”项下方法配制低、中、高质量浓度(厚朴酚2.28、9.10、18.21 μg/mL,和厚朴酚0.78、3.12、6.24 μg/mL,下同)和定量下限质量浓度(厚朴酚1.14 μg/mL、和厚朴酚0.39 μg/mL,下同)的质控样品,按照“2.2.1”项下方法处理后,再按照“2.3”项下条件连续进样测定6次,记录峰面积,考察日内精密度;连续测定3 d,考察日间精密度。以实测质量浓度与理论质量浓度的相对误差(relative error,RE)来表示准确度。结果(表2)显示,定量下限及低、中、高质量浓度质控样品的日内、日间RSD和RE的绝对值均在5%以内,符合药典相关要求[8]。

表2 不同血浆中厚朴酚、和厚朴酚定量分析的精密度与准确度试验结果
2.4.5 提取回收率与基质效应试验 按照“2.1.3”项下方法配制低、中、高质量浓度的质控样品,每质量浓度平行6份。按照“2.2.1”项下方法处理后,再按照“2.3”项下条件进样测定,记录待测成分的峰面积(As)。分别取适量空白血浆,按照“2.2.1”项下方法处理后,加入适量某质量浓度混合工作液,配制成相应质量浓度(与低、中、高质量浓度相同)的待测样品,每质量浓度平行6份,按照“2.3”项下条件进样测定,记录待测成分的峰面积(As’)。按照“2.1.3”项下方法操作,以甲醇代替空白血浆,平行配制6份低、中、高质量浓度的待测样品,按照“2.2.1”项下方法操作后,再按照“2.3”项下条件进样测定,记录待测成分的峰面积(Bs)。提取回收率=As/As’×100%;基质效应=As’/Bs×100%。结果见表3。
表3 不同血浆中厚朴酚、和厚朴酚定量分析的提取回收率和基质效应结果(±s,n=6,%)

表3 不同血浆中厚朴酚、和厚朴酚定量分析的提取回收率和基质效应结果(±s,n=6,%)
待测成分厚朴酚理论质量浓度/(μg/mL)2.28 9.10 18.21 0.78 3.12 6.24提取回收率基质效应牛血浆98.65±0.44 97.98±0.47 98.57±0.33 98.37±0.62 98.05±0.53 98.20±1.04兔血浆96.17±0.32 96.81±0.47 95.74±2.28 97.02±1.03 96.87±0.52 97.19±0.61和厚朴酚大鼠血浆97.77±0.33 98.46±0.48 97.69±0.44 98.67±1.04 98.52±0.53 98.84±0.62牛血浆97.02±3.42 93.18±3.94 97.33±3.63 94.83±2.18 94.03±3.18 95.86±2.57兔血浆97.20±2.97 95.08±2.34 96.51±1.84 95.32±2.45 93.04±2.84 100.28±3.85大鼠血浆98.65±1.46 97.08±1.76 98.05±2.28 95.46±1.48 95.79±1.67 96.97±2.28
2.4.6 稀释可靠性考察 在实际测定时,往往会遇到个别样品质量浓度超过线性范围上限的情况,为了不影响实验的精密度和准确度,本研究向空白血浆中加入适量的混合对照品储备液,配制成厚朴酚、和厚朴酚质量浓度分别为227.60、78.00 μg/mL的样品溶液,用空白血浆稀释10倍后,按照“2.2.1”项下方法处理,再按照“2.3”项下条件进样测定,记录峰面积并计算待测成分的变异系数。结果显示,厚朴酚、和厚朴酚的变异系数分别为1.13%、0.88%,符合药典相关要求[8]。
2.4.7 样品稳定性考察 按照“2.1.3”项下方法配制低、高质量浓度的质控样品,每质量浓度平行6份。考察样品室温放置6 h、反复冻融(-80 ℃~室温)3次、-80 ℃放置6个月、按照“2.2.1”项下方法处理后在自动进样器中放置24 h、按照“2.2.1”项下方法处理后室温放置6 h的稳定性。结果显示,上述样品中各待测成分色谱峰峰面积的RSD均小于1%(n=6),符合药典相关要求[8]。
2.5 平衡透析试验
将透析袋剪成长约8 cm的小段,以0.01 mol/L碳酸氢钠溶液活化后,用水洗净,置于空白透析液中,于4 ℃下保存,备用。取活化后的透析袋,用棉线结扎一端使不漏液,精密量取牛、兔、大鼠血浆1 mL(即透析内液),分别置于透析袋中,将袋口扎紧。将透析袋分别浸没于装有质量浓度为4.5、9.0、13.5 μg/mL含药透析液8 mL的离心管中,调节透析袋内外液面,使两者保持在同一水平并尽量避免透析袋接触到离心管壁,放入37 ℃恒温箱中平衡透析24 h。待平衡透析结束后,取少量透析外液加至10%高氯酸溶液0.5 mL中以检查是否有血浆渗漏(产生白色沉淀者作废)。取透析内液和透析外液,分别按照“2.2.1”“2.2.2”项下方法处理后,再按照“2.3”项下条件进样测定,记录峰面积并按随行标准曲线计算待测成分的质量浓度,并按下式计算血浆蛋白结合率:血浆蛋白结合率(%)=(c内-c外)/c内×100%(式中,c内、c外分别表示透析内、外液中待测成分的质量浓度)。每质量浓度平行3次。使用SPSS 18.0软件对数据进行统计分析,数据以±s表示,多组间比较采用方差分析,组间两两比较采用LSD-t检验,检验水准α=0.05。结果见表4。
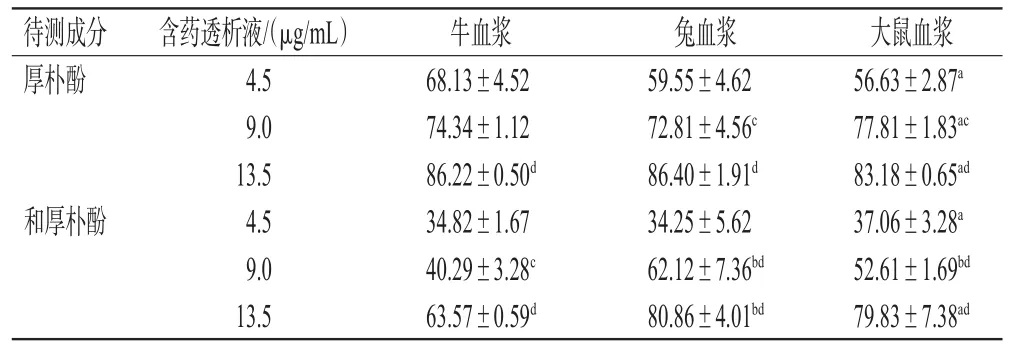
表4 不同血浆中厚朴酚、和厚朴酚的蛋白结合率测定结果(n=3,%%)
由表4可见,在相同种属的血浆样品中,厚朴酚、和厚朴酚的蛋白结合率均随含药透析液质量浓度的增加而升高,有一定的浓度依赖趋势(P<0.05或P<0.01)。在相同质量浓度的含药透析液中,大鼠、兔血浆样品中厚朴酚、和厚朴酚的蛋白结合率与牛血浆样品比较,差异大多具有统计学意义(P<0.05或P<0.01),且在9.0、13.5 μg/mL的含药透析液中,兔血浆样品中和厚朴酚的蛋白结合率均显著高于牛血浆样品(P<0.01)。
3 讨论
3.1 色谱与质谱条件、样品前处理方法优化
本研究应用了UPLC-MS/MS技术,该技术分离效果佳、灵敏度高、稳定性好,具有较高的选择性和特异性,可实现高通量检测[9]。本课题组前期发现,与甲醇-水流动相体系相比,乙腈-水流动相体系的系统压力更低;在水相中加入适量的甲酸,可保证色谱峰峰形较好。本课题组前期比较了待测成分在不同粒径、不同长度及内径的色谱柱上的分离效果,最终选择ACQUITY UPLC BEH C18(2.1 mm×50 mm,1.7 μm)作为色谱柱进行分析。本研究采用了ESI,可用于准确测定待测成分的分子离子峰;此外,本研究进一步对离子传输管温度、气化室温度、去簇电压、碰撞电压等质谱参数进行了优化,最终确定了“2.3”项下色谱与质谱条件。本研究样品前处理采用了蛋白沉淀法,该方法操作简单且重现性好;同时,血浆样品经蛋白沉淀、离心后的上清液以氮气流浓缩富集,可避免待测样品被沉淀剂(乙腈)稀释。
3.2 血浆蛋白结合率检测方法选择及结果分析
药物血浆蛋白结合率的常用研究方法包括平衡透析法、超滤法、超速离心法、高效亲和色谱法、高效毛细管电泳前沿分析法等[10-11]。本研究采用了经典的平衡透析法,该法经济,受外界环境影响小,结果稳定、可靠[11]。本课题组前期研究发现,透析袋吸附率的变异系数均小于1.00%,不影响蛋白结合率的测定;随着透析时间的延长,各待测成分的血浆蛋白结合率亦不断增大,其在相邻时间点(平衡透析24、48 h)的血浆蛋白结合率并无明显差异,故将平衡透析时间设为24 h。同时,参考人体温度,将平衡透析温度设为37 ℃。
含药透析液质量浓度是根据有效剂量下所测血药浓度而定,需覆盖常规剂量下的血药浓度范围[12],故本研究将含药透析液质量浓度设为4.5、9.0、13.5 μg/mL。结果表明,在相同种属血浆中,厚朴酚、和厚朴酚的蛋白结合率均随含药透析液质量浓度的增加而升高;在相同质量浓度的含药透析液中,大鼠、兔血浆样品中厚朴酚、和厚朴酚的蛋白结合率与牛血浆样品有所不同,且兔血浆样品中和厚朴酚的蛋白结合率(9.0、13.5 μg/mL的含药透析液)显著高于牛血浆样品。笔者认为,透析内液和外液间的透析扩散动力来源于透析袋两侧待测成分的浓度差,中、低质量浓度含药透析液中各待测成分与血浆蛋白的结合程度较低且尚未达到饱和,而高质量浓度含药透析液中各待测成分具有较高的扩散动力,故其血浆蛋白结合率较高[13]。同时,厚朴酚与和厚朴酚均为弱酸性成分,多与血浆白蛋白共价结合,在每100毫升牛、兔、大鼠血浆中,分别含白蛋白4.1、3.8、1.68 g[14],提示牛和兔血浆中可能含有更多的结合型成分,这可能是造成2种待测成分蛋白结合率存在种属差异的主要原因。研究指出,人体正常血浆白蛋白一般为35~50 g/L[14],可推测厚朴排气合剂主要药效成分在人血浆中的蛋白结合率较高,临床应用时应避免与其他高血浆蛋白率药物联用,以防止不良反应的发生,但仍需相关研究予以证实。
综上所述,本研究成功建立了测定厚朴排气合剂主要药效成分在牛、兔、大鼠血浆中蛋白结合率的方法,并比较了不同种属血浆蛋白结合率的差异。结果显示,厚朴酚、和厚朴酚在牛、兔、大鼠血浆中的蛋白结合率有明显的种属差异,且有一定的浓度依赖趋势。
