原发性肾小球基底膜相关肾病遗传背景与诊治
2022-12-28匡新宇
匡新宇
上海市儿童医院 上海交通大学医学院附属儿童医院(上海 200062)
肾小球基底膜(glomerular basement membrane,GBM)是由多种带负电荷的糖蛋白聚合物组成的密集凝胶网状结构,是肾小球发育过程中形成的一种特殊的细胞外基质,位于肾小球足细胞和内皮细胞之间,将尿路与血管分隔,对于维持肾小球滤过屏障的正常结构和功能起着至关重要的作用[1]。GBM结构和功能异常可导致多种肾小球疾病,临床表现为血尿、蛋白尿和/或进行性肾功能下降以及多种肾外表现。根据病因不同,GBM相关肾病可分为原发性与继发性两大类,前者主要为编码GBM的主要组分基因变异所致,而后者多为自身免疫性因素或代谢异常所引起。近年来,随着形态学和遗传学技术的不断进展,尤其是透射电子显微镜和下一代测序技术的应用,人们对各类基因变异所致的原发性GBM相关肾病有了更加深入的认识和了解。
1 原发性GBM相关肾病的遗传背景与分类
成熟的GBM 主要由Ⅳ型胶原蛋白、层粘连蛋白(laminin,LM)、巢蛋白(nidogen)、硫酸肝素蛋白多糖(heparan sulfate proteoglycans,HSPG),包括集聚蛋白(agrin)、基底膜蛋白聚糖(perlecan)以及ⅩⅧ型胶原(collagen ⅩⅧ)等组成的[2-3]。GBM的发育具有时空变化和分层组装的特性,在胚胎发育初期,由内胚层细胞向细胞外分泌层粘连蛋白异构体,与细胞表面的受体如整合素、盘状结构域受体(discoid domain receptors,DDRs)、α-抗肌萎缩相关糖蛋白(α-dystroglycan)以及多配体蛋白聚糖结合,形成一种网格样薄膜结构,构成GBM 最初的雏形;后由足细胞分泌Ⅳ型胶原逐渐聚合并沉积在层粘连蛋白聚合体之上,也形成一种网格样结构,两种网格样膜状结构通过HSPG中的agrin和巢蛋白的黏附形成了GBM 的主要组成部分。由此可见,GBM 本质上一种由胶原和层粘连蛋白组成的多层结构,因而人类肾小球基底膜在所有基底膜中几乎最厚(330~460 nm),而大多数基底膜仅有50~100 nm[3]。
GBM 主要结构蛋白异常是导致基底膜相关肾病最重要的原因。成熟的GBM主要包含糖蛋白和胶原两种重要成分。其中,层粘连蛋白是GBM形成的先决条件,作为重要的基底膜糖蛋白,它由ɑ、ß 和γ链3个二硫键连接的多肽组成的16种异构体,层粘连蛋白N端结构域介导了与细胞表面受体如整合素、抗肌萎缩相关糖蛋白等的相互作用,还可进一步与巢蛋白结合而作为连接Ⅳ型胶原网络的桥梁[1,4]。LMβ2的编码基因LAMB2变异可导致皮尔森综合征(Pierson syndrome,PIERS),为一种常染色体隐性遗传,可累及肾脏、眼部及神经系统的综合征,或是主要以肾脏受累而无肾外表现的先天性肾病综合征(NS)[5-6]。Ⅳ型胶原是组成所有基底膜的结构基础,由足细胞分泌,包含由COL4A1-6基因编码的6条α链(α1-6),可组成3种不同的三聚体,分别为α1α1α2、α3α4α5和α5α5α6。业已证实,任何α 3(Ⅳ)、α 4(Ⅳ)、α 5(Ⅳ)和/或α 6(Ⅳ)亚型缺失都会引起Ⅳ型胶原蛋白异质三聚体降解,从而导致GBM 结构异常而致病,如Alport 综合征(Alport syndrome,AS)是由于COL 4 A 3~6的4 个基因变异所致的进行性肾功能降低的遗传性肾脏疾病,还可伴有听力和视力的异常。近年来,随着全外显子测序技术的广泛开展,人们对AS 的遗传背景有了最新的认识,AS分类工作组建议将COL4相关基因变异所致的薄基底膜肾病(thin basement membrane nephropathy,TBMN)和局灶节段肾小球硬化(focal segmental glomerulosclerosis,FSGS)均归类于AS[7-10](表1)。
层粘连蛋白和Ⅳ型胶原蛋白的融合及其与细胞表面受体的结合离不开黏附蛋白、巢蛋白和HSPG 的作用。NID 1基因编码的nidogen-1,可与层粘连蛋白-γ 1 链及Ⅳ型胶原蛋白结合而起到连接基底膜的桥梁作用[1,11]。NID 1基因变异可引起丹迪-沃尔克畸形伴枕部脑膨出(Dandy-Walker malformation and occipital cephaloceles,ADDWOC),表现为小脑发育不全、脑膜异常和枕颅骨缺损,无肾脏表型相关报道,但NID 1基因敲除小鼠模型可出现泌尿系统发育异常[12-13]。HSPG是GBM 负电荷的主要来源,包含agrin、perlecan 以及collagen ⅩⅧ三种主要类型,均在细胞-基质中起黏附作用。其中,编码agrin 或perlecan 的AGRN或HSPG 2基因错义变异可分别导致先天性肌萎缩综合征和Schwartz-Jampel综合征,但并未影响肾功能[2,14-15](表1)。
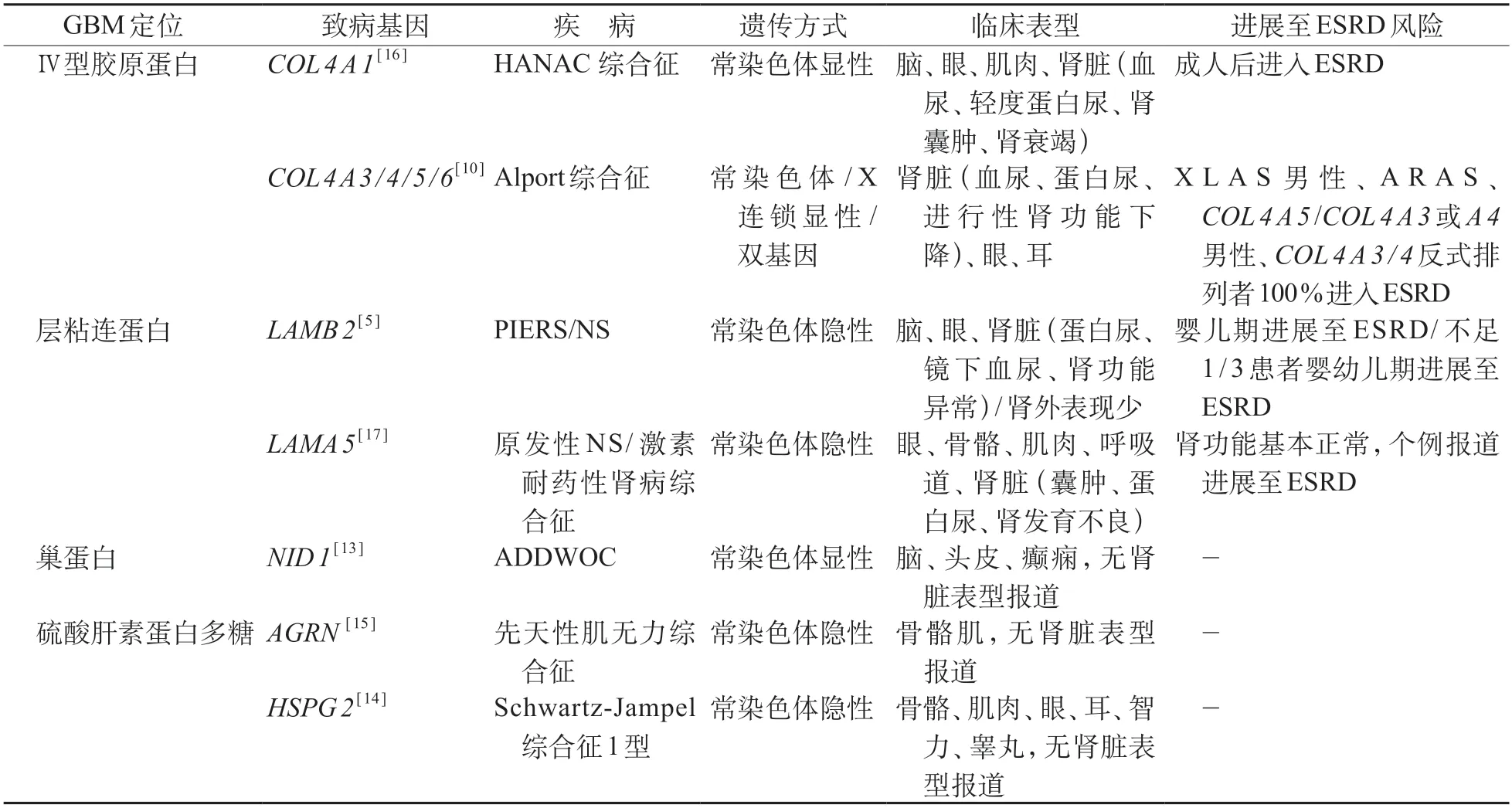
表1 GBM相关肾脏疾病分类、临床表型及预后
由此可见,GBM 主要结构蛋白异常是导致GBM相关肾病最主要的病因,这给临床上判定致病变异的定位信息提供了很好的鉴别要点。
2 原发性GBM相关肾病的诊断要点
原发性GBM相关肾病大都为先天性、罕见性肾脏疾病,发病率低,临床表现各异,缺乏大规模流行病学研究,了解不同GBM相关肾病的诊断要点有助于早期确诊该类疾病。
2.1 血尿
由于GBM 主要结构蛋白异常导致其电荷屏障和分子屏障均受到破坏,血尿几乎成为所有该类肾病的首要表现,早期表现为间歇性或持续性镜下血尿和/或肉眼血尿,根据遗传方式不同各有差异。所有X 连锁显性遗传性AS(ⅩLAS)男性、98%ⅩLAS 女性均可伴有血尿;常染色体隐性遗传性AS(ARAS)与ⅩLAS男性临床表现几乎一致[18]。HANAC 综合征可表现为家族性血尿,患者可出现镜下血尿或发作性肉眼血尿[19];而LAMB 2基因变异所致孤立性肾病综合征可能仅合并轻微镜下血尿表现[20]。
2.2 蛋白尿
蛋白尿也是GBM 相关肾病的主要表现之一。ⅩLAS 男性儿童期即可出现蛋白尿,甚至呈肾病综合征水平;73%ⅩLAS 女性可同时出现血尿伴蛋白尿;ADAS 患者蛋白尿并不少见,但出现时间晚于其他类型,有报道25 例ⅩDAS 患者中有18 例合并蛋白尿,中位出现时间约17岁[18,21]。虽然已报道的COL4A1基因无义变异HANAC综合征病例可表现为血尿合并轻度蛋白尿,但实验研究表明COL 4 A1基因错义变异小鼠模型中出现肾小球延迟发育的现象,新生小鼠可检出大量蛋白尿,但1周后尿蛋白显著减少且不影响成年后肾功能,可见临床症状的变化最终取决于基因变异的方式[16]。大量蛋白尿也是皮尔森综合征和先天性肾病综合征的主要临床表现,患儿生后即可出现严重水肿和低蛋白血症[5-6]。
2.3 肾衰竭
进行性下降的肾功能直至进入终末期肾病(end stage renal disease,ESRD)是影响几乎所有GBM相关肾病预后的关键因素。根据病因不同,患者进入ESRD 的时间也存在很大差异。皮尔森综合征以及先天性肾病综合征患者可在婴幼儿期发生肾衰竭;100%的ⅩLAS 男性通常在40 岁前、100%ARAS 平均21 岁进展至肾衰竭,而仅有15%~30%的ⅩLAS女性在60 岁前发生肾衰竭;COL 4 A 5/COL 4 A 4或COL 4 A 3男性和COL 4 A 3/COL 4 A 4反式排列的双基因变异发展至ESRD 的风险均为100%。多中心基因型与表型研究显示截短变异、大片段缺失及剪切变异等进入ESRD 的中位时间均早于30岁,由此可见Ⅳ型胶原变异对肾脏的损害是巨大的[10,18,22]。但广义上的ADRS患者如果不伴有蛋白尿、FSGS、感音神经性耳聋等危险因素,发生肾衰竭的概率<1%;当合并危险因素时,这一比例将升高至20%[10]。HANAC 综合征患者因多发肾囊肿多在成人后进入ESRD。
2.4 肾外表现
Ⅳ型胶原和层粘连蛋白等广泛存在于多个器官和组织的基底膜中,因此,其结构和功能异常除了引起肾脏表型外,还合并有多种肾外表现。如AS患者可同时伴有感音神经性耳聋、前圆锥样晶状体等眼耳异常[23]。HANAC综合征可伴有脑白质病变、动脉瘤、视网膜动脉迂曲、多囊肾、肌肉痉挛等症状[1]。而PIERS 常合并小瞳孔、神经发育迟缓甚至视力丧失等[20]。肾外表现是诊断GBM 相关肾病的重要线索。
2.5 肾脏病理
在基因检测技术推广前肾活检病理是GBM 相关疾病诊断的金标准,尤其是AS,其典型表现为电镜下肾小球GBM广泛增厚、变薄以及致密层分裂[18];如果没有条件进行电镜分析,免疫荧光检测如发现Ⅳ型胶原ɑ链缺失或连续性中断也可确诊[23];疑似ⅩLAS 患者还可以进行皮肤活检免疫荧光检测,如ɑ 5(Ⅳ)缺失或连续性中断也提示诊断。PIERS 因患儿快速进展至肾衰竭,故肾活检并不作为确诊的必要条件;同样HANAC 综合征因其诸多肾外表现亦不依赖于病理诊断。
2.6 基因检测
随着检测技术的快速发展和应用范围的不断扩展,基因检测已经成为遗传性疾病的必要诊断依据。但因为部分疾病起病时仅表现为镜下血尿和/或微量蛋白尿,对于先开展基因检测还是肾活检病理检测仍然存在很大的争议,不同国家地区的指南推荐方案并不相同。基于活检为创伤性检查,对于肾小球性血尿、有家族史和/或肾外表现的患者可以优先选择基因检测;对于基因检测阴性或变异位点为临床意义未明时建议扩大检测范围,补充病理结果以明确诊断。
当然,该类遗传性GBM肾病应注意与其他临床上表现为肾小球性血尿伴蛋白尿、甚至肾功能异常的肾小球疾病相鉴别,如急进性肾小球肾炎、IgA肾病、溶血尿毒综合征、抗肾小球基底膜病等。伴有肾外表现的还应与其他累及该系统的综合征如眼-脑-肾综合征等相鉴别,以避免误诊漏诊。
3 原发性GBM相关肾病的治疗展望
该类疾病目前均缺乏有效针对原发病的治疗方案。对于PIERS、CNS 等临床肾脏表型严重或HANAC 综合征合并癫痫等肾外症状者应及时给予对症、支持治疗。近年来,随着药物基因组学和基因治疗研究的不断深入,AS 的治疗取得了很大的进展。确诊后建议早期给予血管紧张素转化酶抑制剂(ACEI)和血管紧张素受体拮抗剂(ARB)药物,延缓疾病进展,保护肾功能[24]。现已有4 种药物:甲基巴多索隆、Lademirsen、阿曲生坦,以及笔者所在团队的羟氯喹进入了临床研究阶段[25]。同时,其他治疗方案如干细胞移植、子宫致敏相关基因1(USAG-1)以及针对截短变异基因采用外显子跳跃技术均在AS小鼠模型中显示出强大的GBM修复作用,显著改善了蛋白尿水平[18,26-27]。对于PIERS,有学者采用蛋白质疗法治疗LAMB 2敲除小鼠取得了一定的成效。重组hLM-521三聚体蛋白通过静脉注射入小鼠体内,继而被运送到GBM并停留数周,从而修复GBM的组成,改善足细胞稳态,并延迟蛋白尿的增加,但该治疗方案并不能预防肾病综合征的发生[28]。对于已经进展至ESRD 的患者,肾替代治疗,尤其是肾移植仍是唯一有效的改善预后的治疗手段。
GBM作为肾小球滤过屏障的主要组成部分,连接起内皮细胞和足细胞,通过其复杂的网络结构限制了大分子蛋白及血细胞等的漏出,兼具分子屏障和电荷屏障双重作用。也由此,编码GBM相关蛋白的基因变异所致结构异常是产生多种先天遗传性疾病的重要原因,临床上对于不明原因的血尿和/或肾功能不全的患儿应仔细甄别,基因检测和肾活检病理是确诊疾病的最有效手段。
