水稻立枯病原菌尖孢镰孢菌致病力降低T-DNA突变体的筛选与插入位点鉴定
2022-12-27张俊华杨松润彭莉莉杨明秀徐晓凤李培宁
张俊华,杨松润,彭莉莉,王 亮,杨明秀,徐晓凤,倪 哲,李培宁
(东北农业大学农学院,哈尔滨 150030)
近年来,黑龙江省水稻播种面积逐年增加,但因气温变化大、播种密度大和管理防控措施不到位等情况,黑龙江省水稻立枯病发生严重,秧苗大面积死亡,严重影响水稻品质与产量[1]。
尖孢镰孢菌(FusariumoxysporumSchelcht)是引起水稻立枯病主要病原真菌。该菌可侵染100多种不同植物[2],根据其植物寄主范围,如双子叶植物豆类、单子叶植物香蕉等,表现出复杂的致病特性和遗传多样性[3],被列为世界第5大植物病原真菌[4]。尖孢镰孢菌主要通过分泌细胞壁降解酶和毒素侵害宿主植物[5-6]。细胞壁降解酶负责降解细胞壁,促进尖孢镰孢菌侵入与增殖,其中降解产生的果胶阻塞宿主植物导管导致萎蔫[7]。毒素抑制宿主植物防御反应,促进病菌侵染[6]。Zhang等发现尖孢镰孢菌具有谱系特异性染色体,宿主范围广泛[8]。由于尖孢镰孢菌遗传背景复杂,水稻立枯病防治主要集中在化学药剂上,缺少高效防治方法。认识水稻立枯病菌致病机制是减轻病害重要手段,是寻求水稻秧苗发病有效防治策略的重要突破口[9]。
根癌农杆菌介导的遗传转化(Agrobacterium tumefaciens-mediated transformation,ATMT)自1995年成功转化酿酒酵母,至今已有100多种真菌转化体系构建成功[10]。该方法可使T-DNA随机插入真菌基因组,打断不同位点基因,获得大量突变体。具有操作简单、转化效率高、单次插入概率高的优点,被广泛用于了解宿主与病原物相互作用机制[11]。目前,关于水稻立枯病菌多集中在生物学特性、致病力分化和遗传多样性分析等方面研究,未见水稻立枯病菌致病机理的报道[12]。明确水稻立枯病菌致病力关联基因及功能,深入解析水稻立枯病菌致病分子机制,对农田水稻立枯病防控具有重要意义。
本研究利用ATMT建立尖孢镰孢菌T-DNA突变体库,获得具有对真菌发育有影响和致病力降低的T-DNA插入突变体,定位到突变位点。该信息有助于确定新的致病位点,为深入鉴定基因功能提供研究方向,为水稻立枯病防治提供理论依据。
1 材料与方法
1.1 试验材料
1.1.1 植物材料、菌株及质粒
供试水稻品种为龙粳31;供试菌株为Fo21,东北农业大学植物病理教研室从水稻立枯病株分离获得高致病力尖孢镰孢菌菌株,根癌农杆菌AGL-1和双元载体pBHt2用于尖孢镰孢菌遗传转化,由吉林大学秦庆明教授和张世宏教授馈赠。质粒pBHt2具有潮霉素抗性表达框,试验开始前转入根癌农杆菌AGL-1中备用。
1.1.2 培养基
马铃薯葡萄糖琼脂培养基(Potatodextrose agar,PDA);LB培养基(Luria-bertani,LB);诱导培养基(Induction medium,IM),按张键等方法配置[13]。
1.1.3 主要试剂
抗生素(利福平、卡那霉素、潮霉素B、头孢霉素、羧苄霉素)、乙酰丁香酮、吗啉乙磺酸和其他药剂国产分析纯均购自北京酷来搏科技有限公司;2×TaqPCR Master Mix购自北京康为世纪生物科技有限公司;限制性内切酶EcoRⅠ购自宝日医生物技术(北京)有限公司;地高辛杂交检测试剂盒Ⅱ购自罗氏公司;胶回收纯化试剂盒购自杭州新景生物试剂开发有限公司。
1.2 方法
1.2.1 尖孢镰孢菌突变体构建
参考Mullins等方法进行农杆菌介导遗传转化[14],并稍有改进。将含有双元载体pBHt2的根癌农杆菌AGL-1在含有25μg·mL-1利福平和50μg·mL-1卡那霉素LB固体培养基中划线活化,挑单菌落加入与上述相同浓度抗生素LB液体培养基,28℃,180 r·min-1振荡培养12~16 h。取1 mL菌液7 000 r·min-1离心5 min,弃上清液,沉淀使用IM液体培养基清洗两次,最终加入1 mL含有200μmol·L-1乙酰丁香酮IM液体培养基,28℃,180 r·min-1振荡培养4~6 h。将诱导菌液使用含有200μmol·L-1乙酰丁香酮IM液体培养基稀释至OD600≈0.3,以1∶1体积比将诱导菌液和106个·mL-1分生孢子混匀。取200μL混合液涂布在覆盖无菌玻璃纸的含200μmol·L-1乙酰丁香酮IM固体培养基中,25℃完全黑暗条件下培养2 d。然后取下玻璃纸,转移至含有150μg·mL-1头孢霉素、300μg·mL-1羧苄霉素和80μg·mL-1潮霉素B的PDA固体培养基上,其中150μg·mL-1头孢霉素、300μg·mL-1羧苄霉素用于避免根癌农杆菌AGL-1生长,80μg·mL-1潮霉素B用于筛选尖孢镰孢菌突变体。在25℃培养2 d后揭去玻璃纸,待长出突变菌落后,转移至含有80μg·mL-1潮霉素B的PDA固体培养基。
1.2.2 CTAB法提取病原菌突变体DNA
参考Kuhad等DNA提取方法[15],稍加改进,刮取菌丝用液氮研磨,将其放入1.5 mL离心管中。取650μL预热CTAB缓冲液加入离心管中并于55℃金属浴1 h。加入等量氯仿/异戊醇(24∶1)缓慢摇匀,12 000 r·min-1离心15 min。吸取上清于新1.5 mL离心管中,再次加入等量氯仿/异戊醇(24∶1)缓慢摇匀,12 000 r·min-1离心10 min。吸取上清于新1.5 mL离心管中,加入2.5倍体积预冷无水乙醇,-20℃沉淀1 h,12 000 r·min-1离心15 min,将沉淀使用70%酒精洗涤并在通风厨中吹干残留酒精,30~50μL ddH2O溶解。
1.2.3 阳性突变体PCR鉴定
根据质粒pBHt2的T-DNA片段上潮霉素抗性片段,通过Primer Premier 6设计并使用引物对HPH-F:TCGCCCTTCCTCCCTTTATTTCAG和HPHR:CTACACAGCCATCGGTCCAGAC作PCR验证,检测突变体T-DNA插入片段。PCR反应体系为:5μL 2×Taq PCR Master Mix(Dye)、0.4μL上游引物、0.4μL下游引物、0.5μL基因组DNA,ddH2O补至10μL。PCR反应程序根据康为世纪2×TaqPCRMaster Mix(Dye)商品说明书操作。扩增产物通过琼脂糖凝胶电泳,利用凝胶成像系统拍照分析。
1.2.4 突变体遗传稳定性测定
随机选择10个突变体在不含抗生素PDA固体培养基上25℃培养7 d,并在相同条件下转接培养4次,最后接种在含80μg·mL-1潮霉素B抗性的PDA固体培养基上,根据突变体生长情况判断突变体是否稳定遗传。
1.2.5 致病力测定
参考Klosterman等分生孢子浸根接种方法测定致病力[16],并稍改进,使用打孔器打2个直径为5 mm菌饼加入到PDA液体培养基中,25℃培养3 d;使用无菌纱布过滤,并用血球计数板统计孢子数量,调整至107个·mL-1分生孢子悬浮液;取30 mL 107个·mL-1分生孢子悬浮液接种至水稻幼苗根系,以无菌水为对照。分级标准和病情指数计算公式参照刘金鑫方法[17]。
1.2.6 生长速率测定
使用打孔器打直径为5 mm突变体菌饼接种到PDA固体培养基上,25℃培养7 d,以野生型Fo21为对照,观察菌落生长速度。每个菌株3次重复。
1.2.7 孢子产量测定
使用打孔器打直径为5 mm突变体菌饼接种到PDA固体培养基上,25℃培养10 d,加入5 mL无菌水并使用无菌毛笔刮取分生孢子,使用无菌纱布过滤并用血球计数板测定分生孢子产量。每个菌株3次重复。
1.2.8 Southern blot检测
检测方法按照地高辛杂交检测试剂盒Ⅱ(Roche)操作指南进行。首先,采用CTAB法提取致病力降低突变体DNA,使用EcoR I将基因组DNA酶切,酶切片段通过电泳在1%琼脂糖凝胶上分离,然后转移至带正电尼龙膜上。使用HPH-F和HPH-R引物PCR扩增并作DIG标记,最后将尼龙膜与DIG标记的探针杂交、洗膜和免疫检测。
1.2.9 侧翼序列扩增
利用Hi TAIL-PCR扩增T-DNA插入位点左右两端侧翼未知序列,分别在T-DNA左臂和右臂边界处设计特异性嵌套引物为L1~L3和R1~R3,并与简并引物LAD1~LAD4和接头序列AC1组合进行三轮PCR(见表1)。第一次PCR反应体系为:12.5μL 2×TaqPCR Master Mix(Dye)、1μL特异性引物L1/R1、1μL简并引物LAD、9.5μL ddH2O、1μL基因组DNA。第二次PCR反应体系为:12.5μL 2×TaqPCR Master Mix(Dye)、1μL特异性引物L2/R2、1μL接头序列引物AC1、9.5μL ddH2O、1μL稀释40倍的第一次PCR产物。第三次PCR反应体系为:25μL 2×TaqPCR Master Mix(Dye)、2μL特异性引物L3/R3、2μL接头序列引物AC1、19μL ddH2O、2μL稀释10倍的第二次PCR产物。反应程序参考Liu等扩增方法[18]。

表1 Hi TAIL-PCR引物序列Table 1 Hi TAIL-PCR primer sequence
1.2.10 数据分析
采用IBM SPSS26.0统计软件进行统计分析。采用单因素方差分析,若P<0.05,则差异显著。
2 结果与分析
2.1 尖孢镰孢菌T-DNA突变体构建
根癌农杆菌与尖孢镰孢菌共培养后,通过抗生素筛选,每个平板长出150~200个突变体(见图1A),随机选取10个突变体并利用引物对HPH-F和HPH-R进行PCR验证,发现除野生型Fo21外均扩增出1 000 bp左右片段(见图1B),说明T-DNA片段成功插入尖孢镰孢菌基因组中,构建尖孢镰孢菌T-DNA突变体。

图1 尖孢镰孢菌突变体筛选与鉴定Fig.1 Screening and identification of Fusarium oxysporum mutants
2.2 突变体遗传稳定性测定
在不含抗生素选择压力下培养4代后转移至80μg·mL-1潮霉素B抗性的PDA固体培养基上,25℃培养3 d,以野生型Fo21为对照,发现突变体仍正常生长(见图2)。说明多次转接培养的突变体仍具有潮霉素B抗性,表明T-DNA可稳定插入尖孢镰孢菌基因组中,不随抗生素缺失而丢失,适合用于后续致病力降低突变体的筛选。

图2 突变体遗传稳定性测定Fig.2 Determination of genetic stability in mutants
2.3 致病力降低突变体筛选
从691个突变体中选取34个菌落形态变化的突变体测定致病力(见图3)。以野生型Fo21为对照,有16个突变体致病力显著降低,分别降低15.36%~76.92%。选取其中致病力降低最为严重的6个突变体B9、B92、C36、D65、E17、F78开展后续研究。

图3 致病力降低突变体筛选Fig.3 Screening for mutantswith reduced pathogenicity
2.4 致病力降低突变体菌落形态特征
25℃培养7 d后,观察致病力降低突变体菌落形态变化(见图4)。与野生型Fo21相比,发现突变体B9、C36、D65、E17、F78产生大量气生菌丝,突变体B92、C36、D65、E17菌落色素产生异常,其中突变体B92最终形成纯白色菌落。

图4 野生型菌株Fo21和6个致病力降低的突变体菌落形态特征比较Fig.4 Comparison of colony morphology of wild type Fo21 and six mutants with reduced pathogenicity
2.5 致病力降低突变体生长速率
25℃培养7 d后,测定致病力降低突变体菌落直径(见图5),与野生型Fo21(8.32 cm)相比,4个突变体B9、C36、D65、E17生长速率显著降低,其菌落直径分别为7.10、7.47、5.34、5.51 cm。突变体B92和F78菌落直径分别为8.29和8.27 cm,与野生型菌株差异不显著。

图5 野生型菌株Fo21和6个致病力降低的突变体菌落直径测定Fig.5 Colony diameter of wild type Fo21 and six mutantswith reduced pathogenicity
2.6 致病力降低突变体产孢量分析
25℃培养10 d后,测定孢子产量(见图6)。6个突变体B9、B92、C36、D65、E17、F78产孢数量分别为2.56×107、3.15×107、1.12×107、0.73×107、1.0×107、2.96×107个·mL-1,与野生型Fo21(3.81×107个·mL-1)相比,均显著降低。分别降低32.81%、17.32%、70.60%、80.84%、73.75%、22.31%。

图6 野生型菌株Fo21和6个致病力降低的突变体孢子产量测定Fig.6 Number of conidia of wild type Fo21 and six mutants with reduced pathogenicity
2.7 Southern blot分析
利用Southern blot检测T-DNA插入数量(见图7)。以野生型Fo21为对照,发现突变体B92有2个条带,其余突变体B9、C36、D65、E17、F78仅有一个条带,说明突变体B92有2个T-DNA插入到基因组中,其余突变体均有1个T-DNA插入到基因组。

图7 致病力降低突变体Southern blot检测Fig.7 Southern blot analysis of mutants with reduced pathogenicity
2.8 致病力降低突变体侧翼序列分析
对6个致病力降低的突变体进行Hi TAIL-PCR扩增(见图8)。结果表明,第一轮扩增,特异性较差,几乎无条带;第二轮扩增,特异性较强,获得清晰条带;第三轮扩增,特异性进一步增强,第二轮与第三轮扩增片段相差100 bp左右,与试验设计一致,表明成功扩增T-DNA突变体插入位点的侧翼序列。
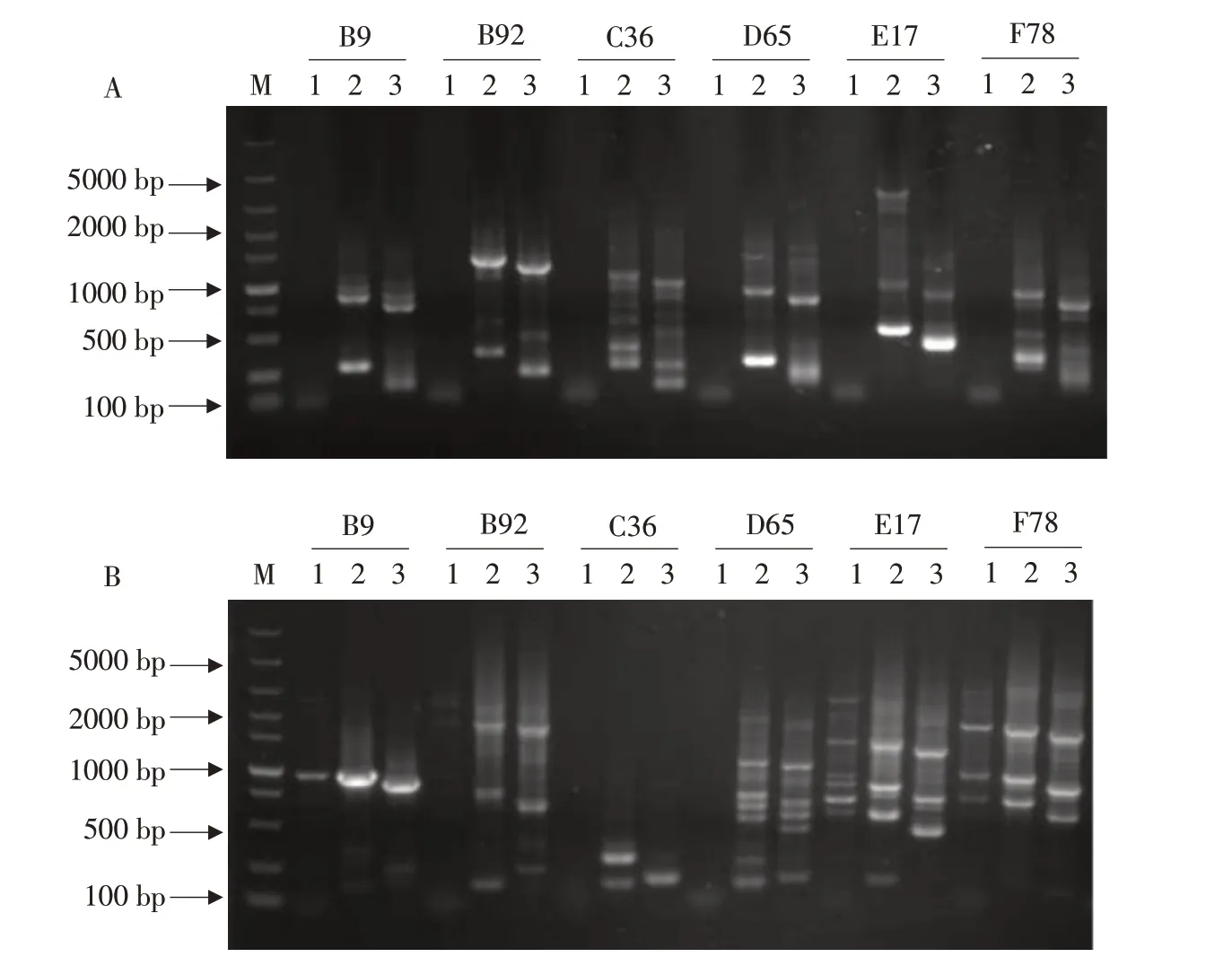
图8 6个致病力降低突变体插入位点侧翼序列扩增Fig.8 Amplification of insertion sites flanking sequence of six mutants with reduced pathogenicity
对上述T-DNA插入位点侧翼序列使用胶回收纯化试剂盒纯化后送至上海生工生物工程股份有限公司获得序列信息,在NCBI基因组数据库中进行BLAST比对分析,确定T-DNA插入到尖孢镰孢菌不同基因位点,主要有3种T-DNA插入位点类型,分别为基因上游、基因编码区和基因下游(见表2)。

表2 6个致病力降低突变体插入位点分析Table 2 Analysis of insertion sites of six mutants with reduced pathogenicity
3 讨论与结论
筛选水稻立枯病菌致病相关基因有助于深入了解和防治水稻立枯病,为发现水稻立枯病新防治手段提供理论依据。因原生质体转化方法受裂解酶、原生质体质量与数量影响,而ATMT克服对原生质体的依赖,孢子、菌丝等均可作为转化材料。近年来ATMT已成为丝状真菌T-DNA插入突变常用手段,可成功筛选致病相关新位点[19-20]。
突变体稳定遗传是后续功能基因注释和生物学特性研究基础,Martínez-Cruz等发现突变体可能发生T-DNA丢失而恢复野生型,与基因组中大量转座子有关。例如,在白粉菌中,突变体仅在有抗生素情况下才稳定存在[21]。本研究成功构建尖孢镰孢菌T-DNA突变体库,对尖孢镰孢菌突变体遗传稳定测试表明,潮霉素B抗性特性可稳定遗传给后代,利用ATMT研究可进一步筛选致病位点、基因功能研究。但尖孢镰孢菌仅从根部侵染,发病周期缓慢,导致土传病原真菌研究滞后。本研究采用分生孢子浸根接种方法筛选致病力降低的突变体,与传统方法相比,症状更明显,发病时间更短[16],最终通过测定34个菌落形态变化突变体致病力,获得16个致病力降低的突变体,选取其中6个致病力降低最为严重的突变体测定生长速度和孢子产量,其突变体菌丝生长和产孢量也发生变化,说明突变体T-DNA插入位点可能影响尖孢镰孢菌生长发育、致病力等方面,因此鉴定6个致病力降低突变体T-DNA插入数量和位点。Southern blot结果表明6个致病力降低的突变体中有一个突变体B92为两个T-DNA插入,其余均为单个T-DNA插入,与Chen等研究T-DNA插入具有高单个插入频率一致[22-24],说明更大概率为单个位点发生突变,可高效筛选导致表型、致病力等变化的基因。目前用于扩增侧翼序列方式较多,如反向PCR、质粒拯救、外源接头PCR、热不对称性PCR等。其中反向PCR受限制性内切酶影响且易形成假阳性片段,质粒拯救虽效率高但操作繁杂,外源接头PCR受限制性内切酶影响且效率较低[25-26]。热不对称性PCR主要是基于特异性引物与简并引物进行高和低退火温度交替循环组成。本次参考Liu等方法加入接头序列,提高热不对称性PCR效率[18]。6个致病力降低突变体的T-DNA插入位点侧翼未知序列被成功扩增,随后在NCBI上作Blast比对,得到插入位点。
本研究首次筛选水稻立枯病菌致病位点,发现使用ATMT可短时间内获得大量突变体,成功筛选到致病力显著降低的突变体,确定T-DNA插入数量和位点,为水稻立枯病发病原因提供更深层次理解,为基因功能研究提供候选条件,为发现水稻立枯病菌新致病机制提供可能。
