N-甲氧基-12-甲酰基红紫素-18己亚酰胺的合成
2022-12-20初晓辉王进军
初晓辉,高 娜,王进军
叶绿素-a 的降解基本都开始于外接环上的羰基反应,进而形成降解产物.例如,人们从海生动物短颈蛤蛎中发现了通过叶绿素空气氧化所得到的叶绿千叶酸-a 甲酯(A)[1],包括132,172-环并脱镁叶绿酸-a 醇(B)、132-羟基叶绿素酮(C)和151-羟基环酮并红质素-18 内酯(D)在内的大量叶绿素降解产物也从不同的海洋生物中分离出来[2].在叶绿素化学研究中,其五元外接环酮的结构转换是衔接和扩展叶绿素碳络的基本手段,可以分别转换成醛基、酮基、内酯和酯基等活性羰基结构,也是合成各种具有叶绿素基本碳架的二氢卟吩衍生物的重要切入点[3-5].
连接于芳香体系上的甲酰基具有极易官能化的化学反应特征,在有机合成中占据特殊位置.对于叶绿素而言,在大环色基的不同位置上引进高反应活性的醛基,是开展化学结构修饰的重要前期工作,也是设计具有应用价值非对称性二氢卟吩四吡咯大环分子(图1)的基本策略.为了深化叶绿素的化学研究,本文基于前期的工作基础,选择脱镁叶绿酸-a 甲酯(1,简称MPa)为起始原料,通过其外接环的转换和空气氧化反应并选择新的温和氧化剂,完成了带有活性甲酰基结构叶绿素衍生物的合成,合成流程详见图2.

图1 具有二氢卟吩基本碳架结构的天然产物
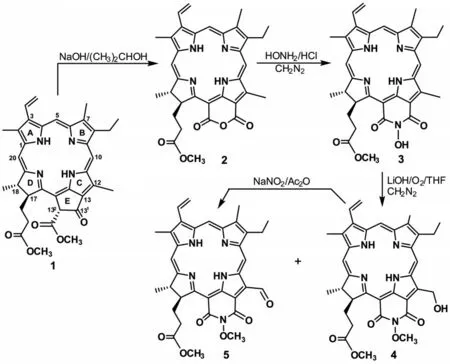
图2 12-含氧基团取代的N-甲氧基-红紫素-18 己亚酰胺的合成
脱镁叶绿酸-a 甲酯1 的五元E-环有两边连接于二氢卟吩母环,为了维持大环自身特定的非平面结构,与外接环共用的两边已经形成定向扭曲,而余下的两个碳原子则分别采取sp3和sp2的杂化形式.由于整个分子的非对称性,外接环很难与大环形成共平面,因此,僵直的刚性赋予了E-环上碳氧双键及其邻位亚甲基以很高的化学反应活性.在E-环羰基的邻位还连有一个含有多氧成分的甲氧甲酰基,所构成的β-酮酯部分应该显示出酮、酯和活泼亚甲基的化学反应活性.除了E-环上在132-位的α-活性氢以外,12-位甲基与E-环酮通过C12-C13双键可以形成烯酮结构,131-位羰基的电子效应可以通过双键的插烯作用而传递到C12-甲基上,因而可以推测出12-位甲基应该具有活性α-位的相应反应性质.将MPa 1 溶解于异丙醇和四氢呋喃的混合溶液中,然后加入氢氧化钠水溶液,在60 ℃条件下,通入氧气剧烈搅拌,以53%的产率生成红紫素-18 酯2,将其在吡啶中与盐酸羟胺作用,历经胺解和再环合过程,高产率地得到N-羟基取代的红紫素-18 酰亚胺3.在氢氧化锂催化下,N-甲氧基红紫素-18 酰亚胺甲酯3在甲醇和四氢呋喃混合液中开口剧烈搅拌,与空气中的氧气顺利地发生氧化反应,然后再于5%硫酸甲醇溶液进行酯化,分别以42%和21%的产率分离出N-甲氧基-12-羟甲基红紫素-18 己亚酰胺4 和目标产物N-甲氧基-12-甲酰基红紫素-18 己亚酰胺5.
为了使得阶段性氧化产物4 的C12-位羟甲基进一步转换成高活性的甲酰基,笔者尝试了包括PCC、活性二氧化锰和琼斯试剂在内的多种氧化试剂,其结果均不理想.最后,将溶解于乙酐中的红紫素-18 亚酰胺用亚硝酸钠快速处理,则以49%的产率得到期待的氧化产物5.
1 材料与方法
1.1 仪器与试剂
使用上海产UV-1900 型紫外分光光度计测定UV-vis,溶剂为二氯甲烷;使用Bruker ARX-400 型核磁共振仪测定1H NMR,溶剂为CDCl3,内标为TMS;使用Perkin-Elmer 1730 型傅立叶变换红外光谱仪测定IR(KBr 压片);使用Perkin-Elmer 2400 型元素分析仪测定元素分析(所有新化合物的C、H、N 的元素分析数值均小于3%).所用试剂均为分析纯或者化学纯,脱镁叶绿酸-a 甲酯按照文献[5]制备.
1.2 红紫素-18 甲酯2 的合成
将1 g(1.649 mmol)脱镁叶绿酸-a 甲酯1溶解于100 mL 异丙醇和200 mL 丙酮中,取7.7 g 氢氧化钠溶于50 mL 水中,并将其倒入混合液中,在空气中开口搅拌2 h 后,用25%的硫酸溶液将pH 值调节为2,先后加入10 mL水和15 mL 二氯甲烷分层,分出有机相,水相用二氯甲烷萃取,合并有机相,用无水硫酸钠干燥后蒸除溶剂,然后用过量重氮甲烷乙醚溶液甲基化,减压浓缩后硅胶柱层析分离[洗脱剂:V(石油醚)∶V(乙酸乙酯)=2∶1],得到370 mg 紫红色固体2(0.874 mmol),产率为53%.m.p.166~168 ℃;UV-vis(CH2Cl2)λmax:396(1.00),422(0.89),519(0.15),698(0.55)nm;1H NMR(CDCl3)δ:-0.19,-1.51(2H,each br s,NH),1.63(3H,t,J=7.4 Hz,8b-CH3),1.82(3H,d,J=7.5 Hz,18-CH3),2.10~2.89(4H,m,17a-H+17b-H),3.22,3.43,3.60,3.66(3H,each s,CH3+ OCH3),3.61(2H,q,J=7.2 Hz,8a-H),4.60(1H,q,J=7.3 Hz,18-H),5.19(1H,dd,J=8.9,2.2 Hz,17-H),6.20(1H,d,J=11.5 Hz,cis-3b-H),6.30(1H,d,J=17.8 Hz,trans-3b-H),8.00(1H,dd,J=17.8,11.5 Hz,3a-H),8.90,9.60,9.65(each 1H,each s,meso-H).IR(KBr)v:2 976~2 850(C-H),1 738,1 695(C=O),1 638(C=N),1 615(C=C)cm-1.
1.3 N-羟基红紫素-18己内酰胺甲酯3的合成
将200 mg(0.357 mmol)红紫素-18 2 溶于15 mL 吡啶中,并向其中加入500 mg 盐酸羟胺,室温下搅拌反应5 h,加入3%盐酸6 mL,水200 mL,用二氯甲烷萃取(100 mL×3),有机相水洗除酸,减压蒸除溶剂后,500~800 目硅胶制备薄层色谱分离[展开剂:V(石油醚)∶V(乙酸乙酯)=1∶1],得到157 mg 紫红色固体3(0.264 mmol),产率78%.m.p.215~218 ℃;UVvis(CH2Cl2)λmax:369(0.72),424(1.00),557(0.27),717(0.40)nm;1H NMR(400 MHz.CDCl3)δ:0.51,0.43(each br s,each 1H,NH),1.60(t,J=7.6 Hz,3H,8-CH3),1.69(d,J=7.3 Hz,3H,18-CH3),2.68~2.80,2.37~2.53,1.88~2.20(each m,4H,17a+17b-H),3.71,3.60,3.29,3.08(each s,each 3H,CH3+OCH3),3.55(q,J=7.6 Hz,2H,8a-H),4.30(q,J=6.8 Hz,1H,18-H),5.18(dd,J=8.4 Hz,1H,17-H),6.16(d,J=11.1 Hz,3b-H),6.26(d,J=17.6 Hz,3b-H),7.83(dd,1H,J=17.6,11.1 Hz,3a-H),9.65,9.11,8.24(each s,each 1H,meso-H);IR(KBr)v:3 430(N-H),2 977~2 886(C-H),1 703~1 738(C=O),1 631(C=C)1 549(chlorin skeleton),1 501,1 420,1 010 cm-1.
1.4 12-羟甲基-N-羟基红紫素-18 己内酰亚胺甲酯4 和12-甲酰基-N-羟基红紫素-18己内酰胺甲酯5 的合成
在35 mL 二氯甲烷中溶解380 mg(0.720 mmol)N-羟基红紫素18 酰亚胺甲酯3,然后,加入氢氧化锂水溶液(0.4 g LiOH+5 mL 水,再加20 mL 甲醇),室温条件下剧烈搅拌4 h,用25%的乙酸将反应液调至pH≈6,加入二氯甲烷和水分层,分出有机相后用二氯甲烷萃取(150 mL×3),将有机相合并,用水洗涤至中性,用无水硫酸钠干燥,过滤后减压浓缩至干.用3 mL 二氯甲烷溶解后加入重氮甲烷乙醚溶液,振摇后(约1 min)快速用冰乙酸处理未反应的重氮甲烷,再用二氯甲烷和水萃取洗涤(150 mL×3),合并有机层,水洗,无水硫酸钠干燥,减压蒸除溶剂,500~800 目硅胶制备薄层色谱分离[展开剂:V(苯)∶V(甲醇)=10∶1],分别得到12-羟甲基-12-去甲基-N-甲氧基红紫素18 乙酰亚胺甲酯4(42%)和12-甲酰基-N-甲氧基红紫素-18 乙酰亚胺甲酯5(21%).4:m.p.291~294 ℃;UV-vis(CH2Cl2)λmax:367(0.57),419(1.00),556(0.25),651(0.10),708(0.37)nm;1H NMR(400 MHz.CDCl3)δ:0.65,-0.02(each br s,each 1H,NH),1.63(t,J=7.6 Hz,3H,8-CH3),1.71(d,J=7.3 Hz,3H,18-CH3),2.75~2.85,2.38~2.54,1.88~1.99(each m,4H,17a+17b-H),4.38,3.59,3.28,3.09(each s,each 3H,CH3+OCH3),3.57(q,J=7.6 Hz,2H,8a-H),4.30(q,J=7.3 Hz,1H,18-H),5.19(d,J=7.3 Hz,1H,17-H),5.78(br s,1H,12-OH),6.03(d,1H,J=15.2 Hz,12a-H),6.07(d,1H,J=15.2 Hz,12a-H),6.17(d,J=11.5 Hz,1H,3b-H),6.27(d,J=17.8 Hz,1H,3b-H),7.81(dd,1H,J=17.8,11.5 Hz,3a-H),9.47,9.16,8.42(each s,each 1H,meso-H);IR(KBr)v:3 430(N-H),2 982~2 888(CH),1 696~1 737(C=O),1 630(C=C)1 548(chlorin skeleton),1 512,1 412,1 023 cm-1.5:m.p.234~237 ℃;UV-vis(CH2Cl2)λmax:379(0.70),423(1.00),585(0.27),659(0.24),715(0.44)nm;1H NMR(400 MHz.CDCl3)δ:0.80,-0.06(each br s ,each1H,NH),1.60(t,J=7.6 Hz,3H,8-CH3),1.67(d,J=7.3 Hz,3H,18-CH3),2.76~2.86,2.48~2.58,2.36~2.45,1.85~1.95(each m,4H,17a+17b-H),4.35,3.62,3.22,2.97(each s,each 3H,CH3+OCH3),3.49(q,J=7.6 Hz,2H,8a-H),4.18(q,J=7.4 Hz,1H,18-H),5.07(d,J=9.1 Hz,1H,17-H),6.16(d,J=11.5 Hz,1H,3b-H),6.22(d,J=17.8 Hz,1H,3b-H),7.68(dd,1H,J=17.8,11.5 Hz,3a-H),10.21,8.79,8.16(each s,each 1H,meso-H),11.78(s,1H,CHO);R(KBr)v:3 429(N-H),2 977~2 892(C-H),1 697~1 743(C=O),1 632(C=C)1 550(chlorin skeleton),1 501,1 422,1 034 cm-1.
在5 mL 乙酐中溶解50 mg 化合物4,室温搅拌下一次性加入研成粉末状的120 mg 亚硝酸钠,继续搅拌1 min,迅速加入10 mL 冰水,用二氯甲烷萃取(10 mL×3),水洗、干燥、浓缩,500~800 目硅胶制备薄层色谱分离[展开剂:V(苯)∶V(甲醇)=10∶1],分离出化合物5(49%).
2 结果与讨论
2.1 红紫素-18 己亚酰胺的氧化反应机理
二氢卟吩3 的外接环结构为环己内亚酰胺,两个内酰胺羰基分别连接在二氢卟吩色基的13-位和15-位上,二者都可以通过插烯结构以共轭的形式与12-位甲基构成一个类同羰基α-位的反应区域.在碱性条件下,氢氧负离子促进了内亚酰胺羰基的烯醇式异构,所生成的烯醇负氧离子3a 通过电子转移与氧分子发生反应,进而夺取反应体系中的质子形成二氢卟吩过氧化氢3b,详见图3.

图3 N-甲氧基-红紫素-18 己亚酰胺的空气氧化反应机理
氢氧负离子可以通过两种形式的亲核作用截断过氧键,由于过氧键的吸电子作用使得C121-碳上带有部分正电荷,所以第一种截断形式是氢氧负离子以亲核取代的方式直接进攻12-位碳原子,作为离去基团的氢氧负离子被羟基取代,得到12-羟甲基红紫素-18 己内亚酰胺4;另外一种是进攻12-位氢原子发生异构化(类似于消去反应),生成连有烯醇过氧键中间体3c,然后,再经电子转移开裂过氧键,去除一个氢氧负离子,形成了碳氧双键,从而转化成12-甲酰基取代的红紫素己内亚酰胺衍生物5.
作为氧化剂的亚硝酸钠A 的氧化过程首先与乙酸酐B 发生亲核加成反应,所形成的四面体中间体C 经电子转移转化成乙亚硝酸酐D 和乙酸根E.乙酰基的引入活化了氮氧双键的反应活性,12-羟甲基红紫素-18 亚酰胺4(分子式中P=Purpurin 红紫素母环色基)可以与混酐E 中的N=O 进行亲核加成而生成中间体F,然后反应体系中的碱(如乙酸根、亚硝酸根和乙酐等)夺取羟基α-位质子,促进完成氧化过程而得到12-甲酰基红紫素-18 亚酰胺5,同时也生成了负氮离子G,后者再经电子转移裂分为乙酸H 和一氧化氮I,详见图4.
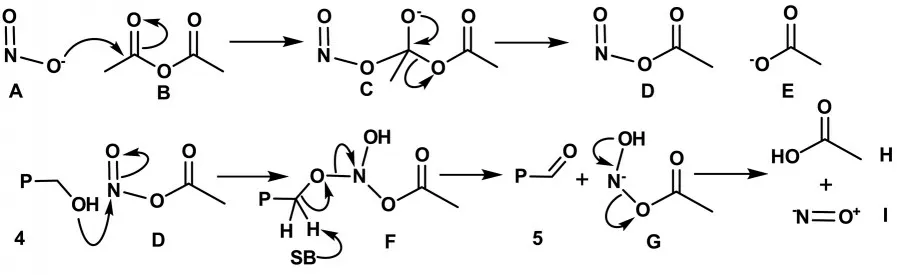
图4 12-羟甲基红紫素-18 亚酰胺4 与亚硝酸钠的氧化反应机理
在不同反应条件下,分别选择PCC、活性二氧化锰和琼斯试剂等专属氧化剂对化合物4 的12-位羟甲基实施氧化,除了活性二氧化锰得到极微量的目标产物以外,其他反应均给出难于处理的粘稠物,其原因可能是二氢卟吩大环的多电子密度区域对氧化剂过于敏感,从而形成混乱的氧化产物.
2.2 红紫素-18 己亚酰胺的12-位结构变化与氢谱表征
N-取代红紫素-18 己亚酰胺3~5 的核磁共振氢谱(图5、图6)清晰地反映出其12-位的结构变化,同时也有力地佐证了作者所提出的氧化反应机理.
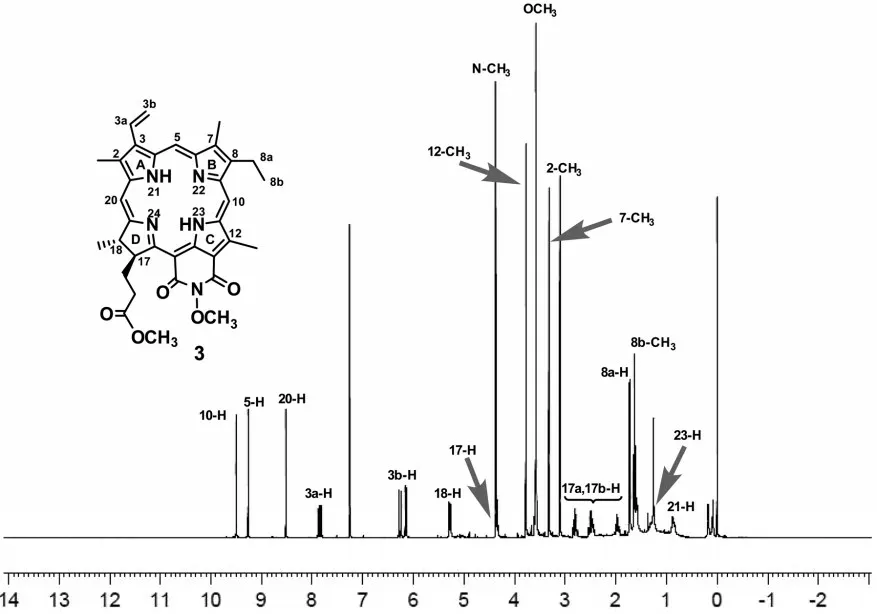
图5 12-羟甲基红紫素-18 亚酰胺3 的1H-NMR 谱图

图6 12-羟甲基红紫素-18 己亚酰胺4 和5 的局部1H-NMR 谱图
在红紫素-18 己亚酰胺整个氧化反应过程中,除了C-亚环上12-位甲基发生变化以外,并没有涉及到其他区域的化学结构.以红紫素-18 几亚酰胺3 的氢谱为例,由于共轭芳香性大环的去屏蔽作用,5-,10-和20-meso-位氢原子的振动吸收均出现在低场(δ>8.00),其A-亚环上乙烯基的3 个氢质子分别出现在δ=7.83、6.26 和6.16 处,顺/反3b-H 和3a-H 分别以大的偶合常数(J=11.1 和J=17.6)相互偶合.C17-位和C18-位的两个质子的化学位移分别裂分为两重峰和三重峰,二者的δ值(5.18 和4.30)也相差较大,其原因可归结17-为质子受到15-位亚酰胺羰基的磁各向异性效应.在δ=3.08~3.71 区域中五个吸收强度很高的3H 单峰隶属于母环上的三个甲基、17-位尾端酯基中的甲氧基和外接环上的N-甲基.8-位上的乙基上两组相互偶合的质子吸收峰则出现在δ=1.69 和δ=1.60 处,二者的偶合常数为J=6.6 Hz.
与化合物3 相比,除了12-位甲基的高强度单峰吸收信号消失以外,红紫素-18 己亚酰胺醇4 和醛5 其他质子的吸收基本不变,12-位连带结构的氢谱变化可以很清晰地证明空气氧化反应过程,也确定了氧化产物4 和5 的化学结构.二氢卟吩醛5 在δ=11.78 ppm 处的低场单峰反映出新生成的12-位甲酰基质子的振动频率,其10-位meso-H 的化学位移也承载着碳氧双键的去屏蔽效应而抵偿移动至10.21 ppm 处;观察二氢卟吩醇4 的核磁共振谱图可以发现,在6.00~6.10 区间出现了裂分为d 峰的两个亚甲基质子吸收峰,在其邻近的δ=5.78 ppm 处凸起一个矮宽的羟基吸收峰,从而确定12-位的甲基已经氧化成羟甲基结构.
3 结论
作为叶绿素-a 的一类重要降解产物,红紫素-18 亚酰胺的六元外接环羰基对12-位甲基具有特殊的活化作用,在氢氧化锂促进下,可以通过插烯效应于12-位上发生空气氧化反应,将甲基分阶段氧化成含氧官能基团羟甲基和甲酰基.通过氧化剂甄选和反应条件优化,最终选择在乙酐中用亚硝酸钠将12-位羟甲基顺利氧化成甲酰基,以理想产率得到所期望的氧化产物,其化学结构均经UV、IR、1H NMR 予以证实.同时,对外接环的结构转换和12-位氧化反应所涉及的相应反应机理进行了研究,为合成高活性醛基取代的叶绿素类二氢卟吩衍生物提供了新的反应途径和理论依据.
