QuEChERS-液相色谱-串联质谱法测定猪肝中玉米赤霉醇含量的不确定度评估
2022-12-20刘曙光隆雪明杨彦宁欧海军陈福华贺宇文
刘曙光 周 灿 隆雪明 杨彦宁 欧海军 张 港 陈福华 贺宇文
(湖南省兽药饲料监察所,湖南 长沙 410006)
玉米赤霉醇,又名“右环十四酮酚”,是玉米赤霉菌正常新陈代谢过程中所产生的次级代谢产物—玉米赤霉烯酮的还原产物,是雷索酸内酯类非甾体类的同化激素。此外,ZAL 还是一种效果良好的皮埋增重剂,具有促进机体蛋白质的合成,提高饲料的转化率和酮体瘦肉率的作用[1-3],被冠以“畜大壮”之称。实验研究表明ZAL在哺乳动物中应用具有一定的危害性[4]。1998 年欧盟就禁止了以ZAL 为代表的激素类药物在畜禽生产养殖上的应用。中国于2002 年发布农业部第193 号公告明确规定禁止ZAL 用于所有食品动物,且所有可食动物源性食品产品中不得检出。2010 年卫生部明确将ZAL 列入动物源性食品中禁止添加使用的药品及其他化合物名单。ZAL 是主要的玉米赤霉烯酮的代谢产物,分别为α-ZAL、β-ZAL,且两者之间可以相互进行转化[5]。李熠等[6]对ZAL 在体内的代谢和毒代动力学进行了较为详细的研究,多达41 种α-ZAL的代谢产物及14 种隐蔽性α-ZAL 的代谢产物首次见诸报道,同时研究指出,α-ZAL 在体内代谢中有效释放出其毒性原型,增加机体暴露的总量且具有显著的种属代谢差异性。此外,该项研究揭示了α-ZAL 致癌性。因此,基于α-ZAL在体内的快速代谢特性和其多种代谢产物总体展现出对动物和人的毒性,及现行检测方法对α-ZAL 不得检出的硬性要求,对《QuEChERS-液相色谱-串联质谱法测定猪肝中玉米赤霉醇》方法进行不确定度评估是十分紧要的。
不确定度,作为一个新颖的质量评价体系术语,它的引入是为了从根本上改变随机误差或系统误差的传统分类,从大数据数理统计角度去测量结果的质量,相比于测量误差等历史统计值来修正测量结果[7-8],主要用于表征合理赋予被测量的值的分散性。测量不确定度被认为是阐明检测结果是否优良的关键性指标[9-11],也是国际上推荐的用于定量评估测量结果的有效手段[12],可分析和评定影响测量结果准确性的各个分量,充分掌握关键因素,提高结果的准确性和可靠性。本文根据JJF1059.1-2012《测量不确定度评定与表示》 和CNAS-GL006-2019《化学分析中不确定度的评估指南》的要求,参照相关文献[13-16],采用QuEChERS- 液相串联质谱法对猪肝中α-ZAL、β-ZAL 的残留进行风险监测,并对得到的不确定度结果进行综合分析和评估,以此来探究影响不确定度的主要因素,为该方法后续在实际检测工作中的应用提供一定的科学依据和理论基础,此外,还可为其它的类固醇类激素药物的残留检测方法的不确定度测量提供一个理论参考。
1 材料与方法
1.1 仪器与试剂
三重四级杆质谱联用仪:QTRAP 4500,美国SCIEX 公司;
超高效液相色谱仪:LC-30A,日本岛津公司;
台式高效冷冻离心机:X1R,德国Thermo公司;
电子天平:PL203,梅特勒- 托利多仪器(上海)有限公司;
固相萃取装置:24TMDL,美国SUPELCO公司;
α- 玉米赤霉醇:100.4 μg/mL,天津阿尔塔β-玉米赤霉醇:100.4 μg/mL,天津阿尔塔乙腈:色谱纯,美国默克;
QuEChERS 萃取盐包:美国Agilent 公司;
Captiva EMR-Lipid:300 mg,3 mL,美 国Agilent 公司。
1.2 实验方法
1.2.1 标准溶液配制
分别用吸量管吸取质量浓度为100 μg/mL的2 种标准储备液0.1 mL 至100 mL 容量瓶中,用乙腈定容至刻度,混匀,即得100 ng/mL 的混合标准中间液。
分别吸取0.1、0.2、0.5、1.0、2.0 mL 混合标准中间液(100 ng/mL) 于空白试样中,余下操作同2.3.2 处理。即得浓度为1、2、5、10、20 ng/mL 混合标准曲线工作溶液。
1.2.2 样品前处理
称取5.00 g 猪肝试样置于50 mL 离心管中,加入3 mL 水和陶瓷均质子,涡旋混匀1 min,加入10 mL 乙腈涡旋提取10 min,加入QuECh-ERS 萃取盐包,混匀,低温离心10 min,取上清液2.4 mL 至10 mL 离心管中,加入0.6 mL水混匀,过Captiva EMR-Lipid(300 mg, 3 mL)脂肪去除柱,挤干小柱,接收全部洗脱液,涡旋混匀,过0.2 μm Nylon 滤膜,滤液供高效液相-串联质谱仪测定。
1.2.3 仪器条件
(1) 色谱条件
色谱柱:Poroshell 120 EC-C8(3.0 mm×100 mm,2.7 μm);柱温:35 ℃;流速:0.3 mL/min;流动相A 为2 mmol/L 乙酸铵0.05%乙酸水溶液,流动相B 为甲醇,(梯度洗脱程序见表1)。流速为0.3 mL/min,柱温:35 ℃;进样量:5 μL。

表1 梯度洗脱程序
(2) 质谱条件
离子源:电子喷雾离子源(ESI),扫描方式:负离子扫描;检测方式:多反应监测(MRM);电喷雾电压(IS):-4500 V;离子源温度(TEM):550 ℃;气帘气(CUR)∶10 psi;雾化气(GS1):50 psi;辅助加热气(GS2):50 psi;碰撞气(CAD):Medium;质谱扫描参数见表2。

表2 质谱参数
1.2.4 残留量计算公式
被测组分残留量按以下公式计算:
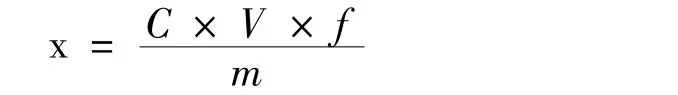
式中:X—试样中被测组分的含量,μg/kg;
C—试液中被测组分的浓度,μg/L;
V—萃取溶剂乙腈体积,mL;
f—试样处理过程中的稀释倍数(本方法为1.25);
m—试样的称样质量,g。
2 不确定度分析与评定
2.1 不确定度来源分析
根据检测流程和残留量计算模型分析,影响试样中α-ZAL,β-ZAL 测定结果不确定度μrel(X)的主要因素有:
(1)待测液的浓度C 引入的相对标准不确定度μrel(C);
(2) 试样称量m 引入的相对标准不确定度μrel(m);
(3)试样前处理V 引入的相对标准不确定度μrel(V);
(4)测量回收率引入的不确定度μrel(R);
(5) 测量重复性引入的相对标准不确定度μrel(χ)。
2.2 不确定度的评定
2.2.1 待测液的浓度C 引入的相对标准不确定度μrel(C)
分析整个实验过程可知不确定度主要来源为标准溶液的配制和标准工作液曲线的拟合等过程。而标准溶液的配制主要包括标准储备液的配制、稀释和标准工作曲线制备等过程。
(1)储备液引入的相对标准不确定度μrel(C1)
由标准物质证书可知,α-ZAL 标准溶液纯度为99.9%,不确定度为±5%,采用均匀分布处理,则因纯度引入的不确定度为
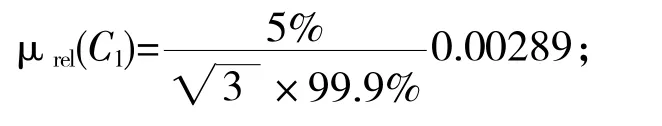
由标准物质证书可知,β-ZAL 标准溶液纯度为99.9%,不确定度为±5%,采用均匀分布处理,则因纯度引入的不确定度为
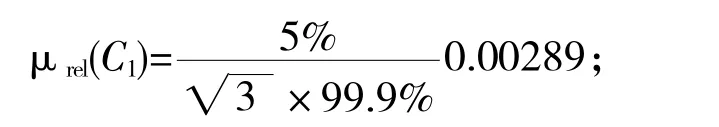
(2)混合标准中间液的配制引入的相对标准不确定度μrel(C2)
用分度吸量管吸取0.1 mL 标准混合储备液至100 mL 容量瓶中,乙腈定容至刻度,混匀,即100 ng/mL 的混合标准中间液。根据常用玻璃量器规定,0.1 mL A 级分度吸量管、100 mL A级单标容量瓶及温度波动引入的不确定度见表3。采用均匀分布,量具及温度波动引入的不确定度见表3。
如表3,标准中间液引入的不确定

表3 标准中间液配制引入的不确定度

(3)标准曲线配制过程引入的相对标准不确定度μrel(C3)
标准曲线配制:分别用0.5 mL、1.00 mL、2.00 mL 吸量管(A 级)分别吸取混合标准中间液(100 ng/mL) 0.1、0.2、0.5、1.0、2.0 mL 于5 个空白试样中,其余操作2.3.2 处理,即得1、2、5、10、20 ng/mL 的标准工作溶液。
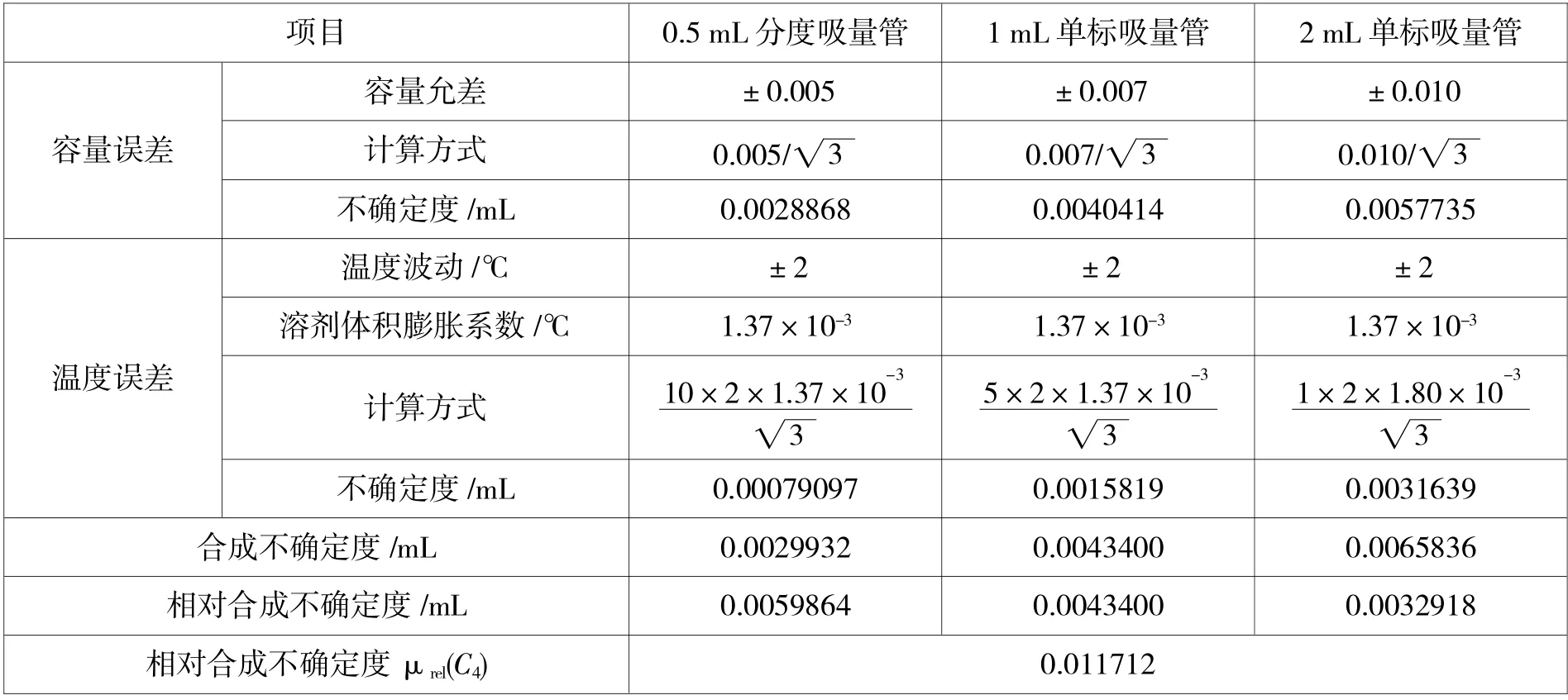
表4 标准曲线配制过程引入的不确定度
如上所示,ZAL 标准曲线配制过程引入的不确定度:

(4)标准曲线拟合引入的相对标准不确定度μrel(C4)
本实验采用外标法定量,标准系列为5 个点,每个浓度点测定2 次后以标准工作液浓度为横坐标,相应浓度的峰面积为纵坐标绘制标准曲线,结果见表5。
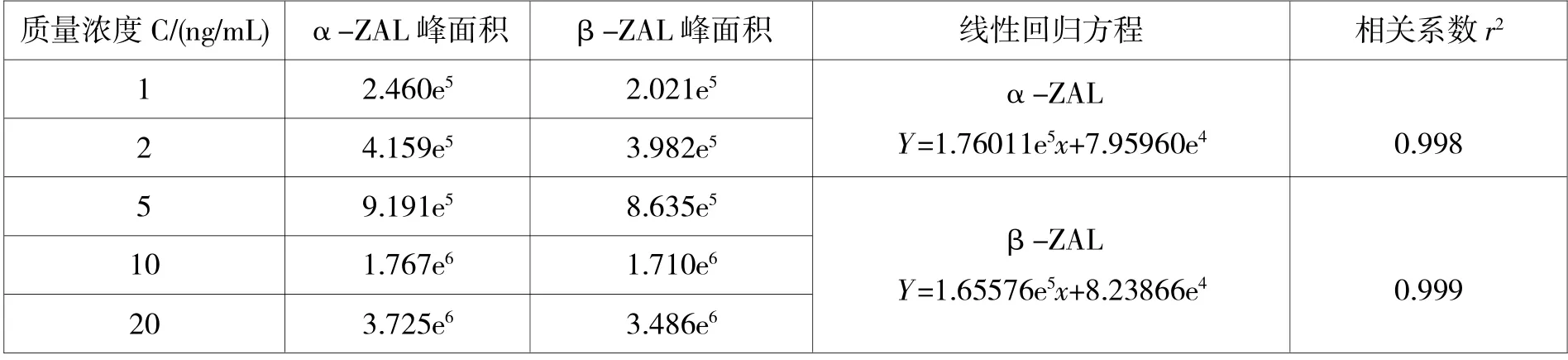
表5 标准曲线
标准溶液峰面积比值残差的标准差,由公式
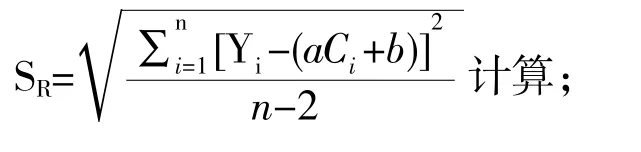
式中p 为对C0测定次数,p=5;n 为标准溶液测定次数n=10;为5 个标准溶液的平均浓度,样品测定5 次后得到的平均浓度结果为C0(见表8),各相应的数值及计算结果见表6。

表6 线性回归方程拟合引入的不确定度
综上所述,α-ZAL 待测液的浓度C 引入的相对标准不确定度μrel(C)

同理可得,β-ZAL 待测液的浓度C 引入的相对标准不确定度μrel(C)=0.04303
2.2.2 试样称量引入的相对标准不确定度μrel(m)
试样称量的相对标准不确定度主要来源于分析天平的校准和样品称取误差。称量采用千分之一天平,由天平检定证书可得,天平的示值允差为±0.005 g,按均匀分布计算,千分之一天平相对标准不确定度为μ(m1)==0.0028868;样品称量5 次,分别为5.012 g、4.988 g、5.006 g、4.994 g、5.000 g,平均称重5.000 g,其标准偏差为0.009482 g,相对标准不确定度为μ(m2)=所以,样品称量的相对标准不确定度为

2.2.3 试样前处理引入的相对标准不确定度μrel(V)
提取过程需要加入10 mL 乙腈涡旋提取10 min,加入QuEChERS 萃取盐包离心,混匀,低温离心10 min,取上清液2.4 mL 至10 mL 离心管中,加入0.6 mL 水混匀,过Captiva EMR-Lipid (300 mg,3 mL)脂肪去除柱,挤干小柱,接收全部洗脱液。其不确定度见表7。
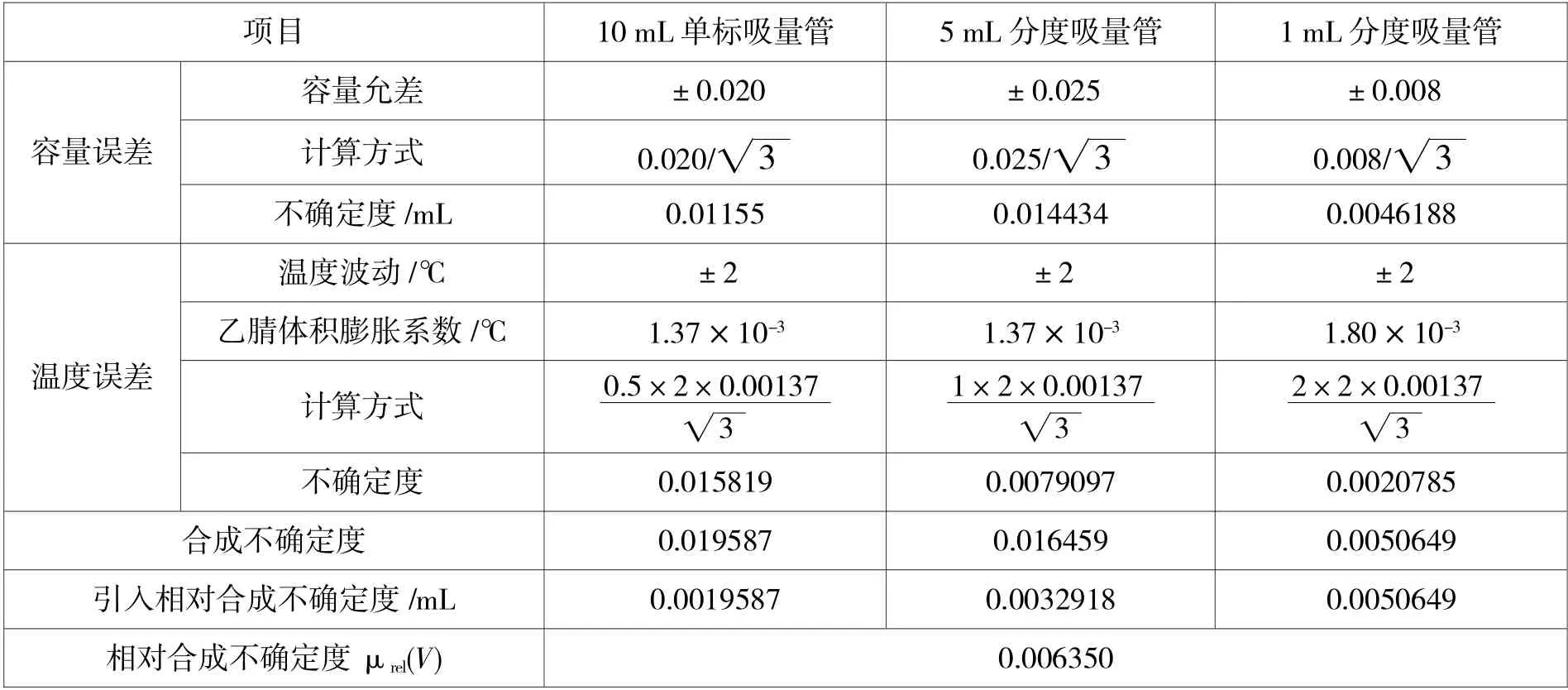
表7 提取过程引入相对合成不确定度μrel(V)
综上所述,ZAL 提取过程引入相对合成不确定度:
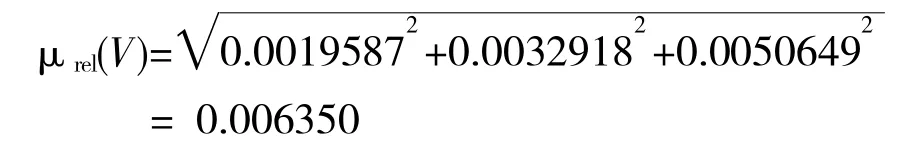
2.2.4 测量重复性引入的相对标准不确定度μrel(χ)
本实验对样品含量为10 μg/kg 的样品进行5 次重复测定,平均含量为(),标准偏差s(x)=标准不确定度为样品重复引入的相对标准不确定度μrel(χ)=测得结果(见表8)。

表8 重复性实验引入的不确定度μrel(χ)
2.2.5 添加回收率引入的相对标准不确定度μrel(R)
对添加水平为10 μg/kg 的样品进行5 次平行重复(n=5),α-ZAL 和β-ZAL 加标回收率引入的相对不确定度结果见表9。
根据JJG 196-2006《测量不确定度评定和表示》需要校正系统误差,因此进行t 分布(公式:,μ 期望值,为平均回收率,n=5,s(R)回收率标准偏差考察平均回收率是否具有统计学意义。若t 计t95=2.57,说明显著差异具有统计学意义,需用回收率校正因子校正测定结果X,否则不必校正。由表9 中可知猪肝中α-ZAL 和β-ZAL 的阳性添加结果都不需校正。
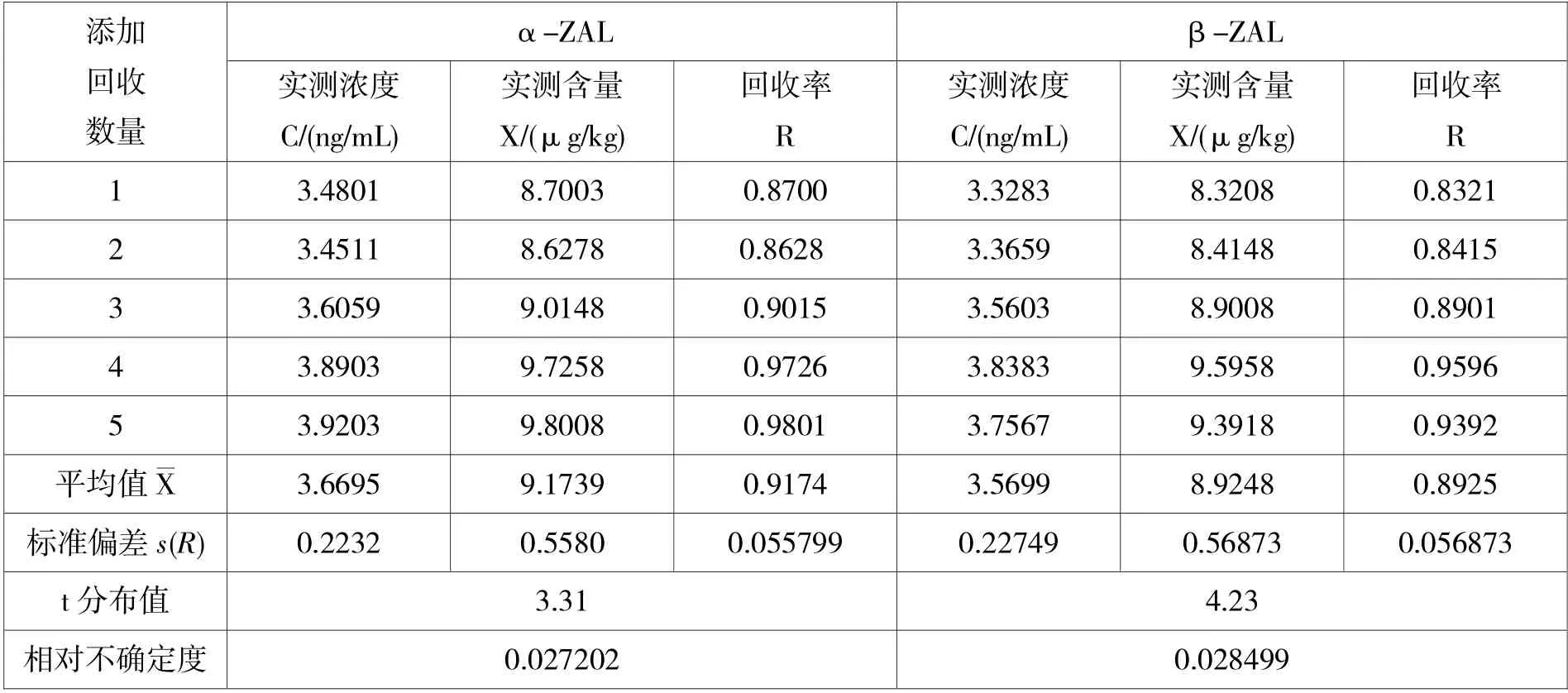
表9 添加回收率引入的相对不确定度
2.2.6 合成相对标准不确定度及贡献率
根据各分量评定的相对不确定度,合成猪肝中α-ZAL 和β-ZAL 测定相对标准不确定度,并评定各个分量对测定不确定的贡献率,各影响因素的不确定结果见表10。合成标准不确定度μrel(X)按下式计算:
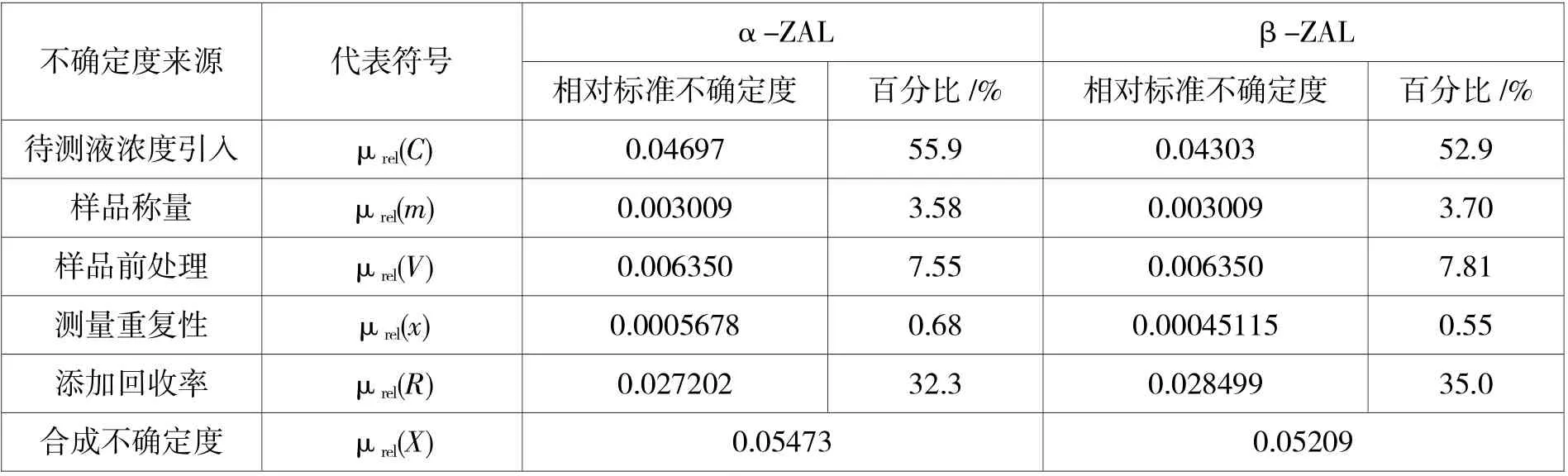
表10 不确定度评定结果
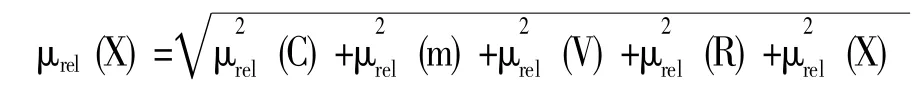
2.2.7 扩展不确定度的计算
化合物扩展不确定度U 根据如下公式计算:

在置信概率为95%,取扩展因子k=2,猪肝中的α-ZAL 和β-ZAL 的扩展不确定度及结果见表11。

表11 不确定度评估结果
根据各不确定度分量的评定结果,待测液浓度引入的不确定度分量最大,其次为添加回收。待测液浓度引入又分为标准储备液的配制和稀释,标准曲线的配制,标准曲线拟合等4 个过程带来不确定度。其中,占比最大的是标准曲线的拟合,其次为标准曲线的配制,评价结果与王同珍等研究[17-18]基本一致,但又存在一些差异,这与采用前处理方法,选用仪器与器皿精密度及考虑因素的多少均相关。
为完善该方法的质量控制体系,在检测过程中可加强一下几个环节的控制而减小不确定度的引入。第一,标准溶液配制主要与量器误差、量器使用频次,以及实验人员的操作水平有关[19-20]。量具的选用,应考虑其误差的大小,合理利用,减少逐级稀释过程;第二,标准曲线拟合带入的不确定度较大,主要是曲线拟合时所选用的C0值的大小决定。在其他条件不变的情况下C0值越大,其曲线拟合的不确定度越小。因而检测中应根据待测样品实际含量和仪器的检测性能,选择合适梯度浓度的标准工作曲线,可降低其不确定度。如有需要,可采取适当浓度单点校正的方式进行计算,亦可大大降低检测过程中的不确定度;第三,方法添加回收是检测过程中质量控制的重要指标,可通过优化前处理过程、规范试验操作等方法提高加标回收率,减小其平行误差,减小测量不确定度。
3 结论
本文基于内部建立的QuECHERS-液相色谱-串联质谱法测定猪肝中玉米赤霉醇含量的方法,分别从待测液浓度、样品称量、样品前处理等方面,对不确定度进行了评价,结果显示影响不确定度的主要因素是标准溶液的配制、标准曲线拟合和方法的添加回收。因此,针对待测浓度及操作过程中等对不确定度影响较大的因素,在检测检验应用中,需要加强控制,以有助于进一步提高测定结果的可信度。
