基于酸位点增强的CeO2掺杂WO3复合催化剂用于柴油机尾气脱硝
2022-12-14牟海峰顾青华
烟 征 ,牟海峰 ,刘 阳 ,顾青华 ,可 欣
(沈阳航空航天大学 能源与环境学院 辽宁省清洁能源重点实验室, 辽宁 沈阳 110136)
近年来,汽车、内燃机尾气中的氮氧化物(NOx)导致了一系列环境问题,例如酸雨、光化学烟雾、臭氧层损耗[1-3]。其中,柴油车数量逐年增加,其排放的氮氧化物所占比重也越来越大。为应对日益严峻的排放问题,中国公布了最新的国六尾气排放标准[4],与之前的国五标准相比,氮氧化物限值降低了66%,达到35 mg/km。氮氧化物选择性催化还原(NH3-SCR)作为柴油车尾气中NOx去除的主要技术之一,具有技术成熟、脱硝效率更高的优点。该技术的核心在于催化剂,常见的催化剂为分子筛以及金属氧化物催化剂。分子筛催化剂中Cu基沸石类[5]效果好,但抗积炭能力弱、易失活[6],并且低温段(150-200 ℃)脱硝效率大多不足60%[6,7],在实际应用过程中遇到了极大挑战。金属氧化物催化剂中的典型代表钒基催化剂(V2O5/TiO2[8]、V2O5¯WO3/TiO2[9]),在国五标准下被认为是最佳商用SCR催化剂[10],但存在操作温度窗口较窄(320-450 ℃)、有生物毒性等缺点[11]。相比燃煤锅炉,柴油车所产生的尾气温度一般为200-400 ℃[12],导致主流商用脱硝催化剂活性温度窗口较窄的问题凸显。
NH3-SCR催化剂的低温(<200 ℃)[13,14]、中温(250-350 ℃)[15]脱硝性能主要受催化剂氧化还原能力及其表面酸位点的影响。Lewis酸位点数量[10,16,17]以及表面活性氧含量的增加[10]有助于促进催化剂低温段“快速SCR反应”[10]的进行,提高脱硝性能。但关于酸位点在中温段的作用机制仍存在争议。例如,有研究指出在中温段SCR反应中,Brønsted酸位点吸附的 N起到主要作用[7],通过吸附NH3形成中间体促进了氮氧化物的脱除。但根据Liu等[18]以及李永龙[19]的研究,中强酸(250-450 ℃)主要促进催化剂中温段SCR反应,即Lewis酸位点在中温段起主要作用。
为同时满足催化剂的宽温域脱硝性能及反应机理探索的需求,本研究选用金属氧化物CeO2以及WO3作为主要活性组分进行复合。CeO2含有弱酸以及中强酸位点,而WO3主要为中强酸位点以及强酸位点[10]。此外,Ce3+和Ce4+之间的相互转换为氧化还原反应的发生提供了丰富的氧空位[20]。WO3作为助催化剂可以改善催化剂表面活性组分的热稳定性,还能通过协同作用提高脱硝效率[21]。目前,已有部分研究将CeO2与WO3用于柴油机尾气脱硝。Shan等[22]制备了CeO2-WO3催化剂,发现两者复合后的脱硝性能相较单组分有显著提升,但并未对脱硝反应机理进行深入探讨。Liu等[10]对CeO2-WO3/TiO2催化剂的反应机理进行了探究,发现CeO2与WO3的复合不仅改善了低温SCR的活性,还形成了更多的Brønsted酸位点,对高温SCR活性和N2选择性有利。
然而,目前多数研究制备出的粉末状催化剂[23]需要继续加工为整体式催化剂,步骤相对繁琐,不利用实际工程应用。考虑柴油车尾部加装脱硝装置的实际条件,不难发现,改善整体式催化剂制备方法对烟气脱硝技术的产业化应用提供了基础。在基底上原位沉积不仅可以简化制备过程,还可以暴露更多的活性位点[24]。常见方法有电化学沉积以及磁控溅射等[25,26],其中,电化学沉积具有成本低、对基材尺寸限制较少、操作简单等优势[16],能够应用于复合金属氧化物催化剂的制备。
综上所述,本研究基于电化学沉积与水热法,以钛网为基底原位负载了CeO2-WO3双活性组分催化剂,将其应用于柴油机尾气的脱硝。研究了电沉积CeO2时间(10、20、40 min)对催化剂性能的影响。围绕酸位点的调控,对所制备的复合催化剂进行脱硝机理研究,通过SEM、EDS、XRD、XPS、H2-TPR、NH3-TPD、原位红外等手段进行表征,探索了CeO2与WO3复合后催化剂酸性位点对脱硝反应的影响规律,获得了反应机理与路径。
1 实验部分
1.1 催化剂的制备
1.1.1 样品预处理
基底为商业钛网(200目,购自河北展墨金属材料有限公司)。首先将样品加工成正方形(1 cm×1 cm),用2 mol/L盐酸、无水乙醇、去离子水分别超声处理15 min,之后放入烘箱内110 ℃下烘干2 h备用。
1.1.2 单一金属氧化物催化剂制备
所有电沉积实验均在CHI660e电化学工作站上进行,方法均为恒电压沉积。电解液制备步骤[27]为:将0.1 mol/L二水合钨酸钠溶液与0.1 mol/L过氧化氢充分混合(pH = 11),然后将0.3 mol/L硝酸加入到混合溶液中(pH = 1.56)。使用银、氯化银/氯化钾作为参比电极,铂网(2 cm × 2 cm)被用作对电极。恒定-0.7 V电沉积20 min,之后在室温下干燥1 h备用。
在电沉积WO3钛网的基础上进行水热负载,水热溶液制备步骤[24]包括:称取钨酸钠二水合物(6.25 mmol)溶于去离子水,加入盐酸水溶液(2 mol/L),然后称取17.5 mmol的草酸以及47.7 mmol的硫酸铵,加入混合溶液中。将混合溶液稀释至125 mL,搅拌30 min后转移至水热反应釜,加入已电沉积WO3的钛网。水热反应在180 ℃下进行16 h,反应完成后用去离子水冲洗三遍钛网,烘干后冷却至室温,记为W-DS。制备好的样品用于后续改性及机理分析。
1.1.3 复合金属氧化物催化剂制备
在单组分催化剂(W-DS)基础上负载CeO2,电解液[28]为0.05 mol/L硝酸铈和0.1 mol/L硝酸钠。首先用O2(6 mL/min)饱和电解液30 min。使用饱和甘汞电极作为参比电极,铂网为对电极。沉积电压-0.8 V,电沉积时间20 min,记为DCe(20)-W。调整电沉积CeO2时间,10 min样品标号DCe(10)-W,40 min样品标号DCe(40)-W。
1.2 催化剂脱硝活性性能的测试
催化剂脱硝性能实验在SCR固定床反应装置上进行。将适量钛网样品放入内径6 mm的石英管中。模拟柴油机尾气环境,烟气总流量为100 mL/min,空速(GHSV)为70000 h-1,烟气组成为:NO(0.05%)、
NH3(0.05%)、O2(3%)、SO2(0.02%)、H2O(5%),N2为平衡气体。SCR反应测试温度200-450 ℃。各反应温度段保持30 min达到反应平衡状态后使用烟气分析仪(Testo Pro350)分析出口气体中的NOx含量。
根据稳态下的气体含量,NOx转化率根据下式计算:

N2选择性根据下式计算:
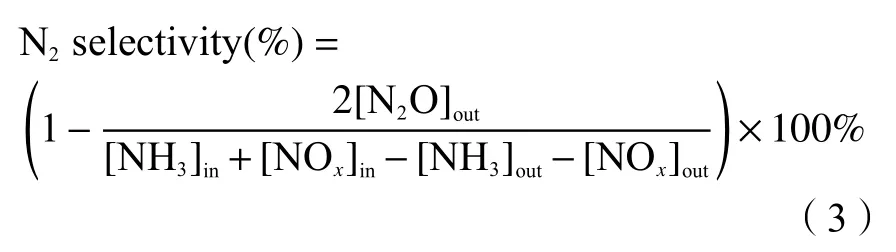
式中,[NOx]out、[NOx]in分别代表NOx的出口质量浓度和进口质量浓度,[NH3]out、[NOx]in分别代表NH3的出口质量浓度和进口质量浓度,[N2O]out代表N2O的出口质量浓度,单位均为mg/m3。
1.3 催化剂的表征
样品形貌与元素组成采用扫描电子显微镜(SEM、EDS,SU-70型)进行分析,加速电压:10 kV,工作距离:6 mm。
采用RINT2000型X射线衍射分析仪进行晶体结构分析(XRD),使用Cu靶Kα射线。扫描速率为4(°)/min,20°-80°扫描,工作电压40 kV,管电流40 mA(12 kW)。
X射线光电子能谱(XPS)使用Thermo Scientific K-Alpha仪器以分析催化剂表面活性组分的化学价态。使用AlKα(1486.7 eV)作为辐射源,功率为225 W,工作电压15 kV,发射电流15 mA,碳(内标)284.8 eV。
NH3程序升温脱附(NH3-TPD)与H2程序升温还原(H2-TPR)使用Bel Cata II仪器进行。NH3-TPD检测中,催化剂在300 ℃下高纯He中预处理30 min,待冷却至80 ℃,以100 mL/min通入NH3(0.1%)/He并保持60 min,然后10 ℃/min升温到700 ℃,记录NH3脱附量。H2-TPR的预处理与NH3-TPD一致,测试在4% H2的氩气流中进行,流量为100 mL/min,以10 ℃/min的加热速率升温至1000 ℃,记录反应数据。
原位漫反射傅里叶红外光谱(in situDRIFTS)分析采用Thermo fisher Nicolet iS50红外光谱仪。扫描分辨率4 cm-1,累积扫描64次。将催化剂置于氩气氛围(50 mL/min),300 ℃下吹扫30 min后降温至250 ℃并记录背景光谱。之后,将样品暴露于不同气氛条件(NH3、NO+O2、NH3+NO+O2)以获得光谱图(去除背景)。
2 结果与讨论
2.1 催化剂脱硝性能测试
2.1.1 不同CeO2电沉积时间对催化剂性能的影响
图1对比了单组分WO3催化剂(W-DS)与不同电沉积CeO2时间的CeO2-WO3复合催化剂(DCe-W系列)的脱硝性能。由图1可得,单组分WO3样品(W-DS)的脱硝效率在200 ℃为62.83%。随着检测温度不断升高至450 ℃,脱硝效率稳定在60%-70%。与单组分催化剂相比,双组分催化剂脱硝效率提升明显。CeO2电沉积时间为10 min的样品(DCe(10)-W)在200 ℃脱硝效率与W-DS样品接近,250 ℃后迅速提升到93.24%,在350 ℃时达到最高值100%,但温度进一步升高后脱硝效率逐步下降,在450 ℃时降至61.48%。电沉积CeO2时间提升至20 min,样品(DCe(20)-W)表现出双组分催化剂最优脱硝性能。温度在200 ℃时脱硝效率显著提升达到91.89%,250 ℃时即达到峰值100%并持续保持到350 ℃,之后脱硝效率逐渐下降,最终在450 ℃时降至70.27%。相较DCe(10)-W,DCe(20)-W样品的活性窗口更宽(200-400 ℃),脱硝效率明显提高,表明CeO2电沉积时间的延长提高了CeO2的负载量,提供了更多的活性位点。然而,电沉积时间延长至40 min,催化剂(DCe(40)-W)的脱硝表现不佳。相较于单组分样品,脱硝效率仅在250-400 ℃略有提升,最高值仅为68.91%。可见,CeO2的沉积可改善WO3的脱硝性能,但过量沉积反而抑制脱硝反应。由称重实验得到电沉积40 min CeO2样品CeO2质量分数变低(10 min-18.03%、20 min-33.33%、40 min-5.66%),这可能是因为过高的CeO2掺杂量导致与WO3的黏附性较差,WO3与CeO2之间的晶格失配增大了界面张力[29]。此外过量掺杂还可能导致离子的传输受阻,WO3纳米棒表面的活性位点被占据[30]。为了明确脱硝性能差异的产生原因,对双组分样品DCe(20)-W及单组分样品W-DS的理化性质及反应机理进行了系统分析与探讨。
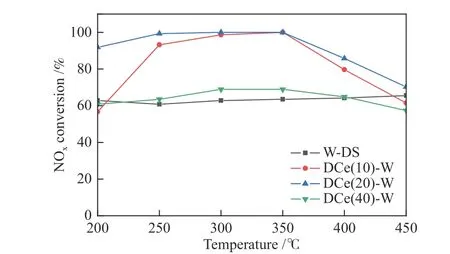
图 1 不同电沉积CeO2时间CeO2-WO3/Ti催化剂脱硝效率Figure 1 Denitration efficiency of CeO2-WO3/Ti catalysts at different electrodeposition time of CeO2
图2对比了单组分WO3催化剂(W-DS)与不同电沉积CeO2时间的CeO2-WO3复合催化剂(DCe-W系列)的NH3转化率以及N2选择性。由图2(a)可知,单组分及双组分样品NH3转化率基本与NOx转化率成正比,变化趋势大致相同。由图2(b)可知,所有样品的N2选择性都有随温度升高而增加的趋势,其中,单组分W-DS样品对应N2选择性最差,双组分改性能够有效提高N2的选择性。
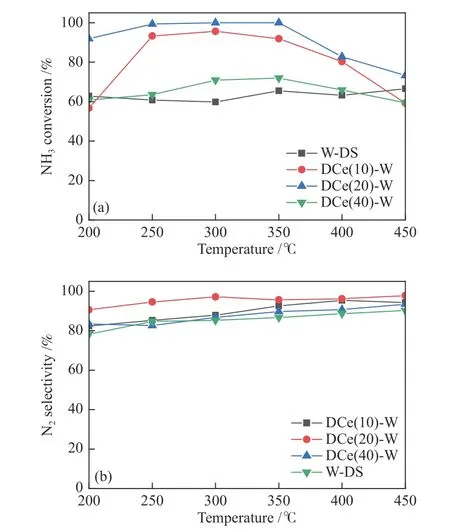
图 2 不同电沉积CeO2时间CeO2-WO3/Ti催化剂脱硝性能Figure 2 Denitrification performance of CeO2-WO3/Ti catalysts with different electrodeposition time of CeO2(a): NH3 conversion; (b): N2 selectivity
2.1.2 催化剂的抗水抗硫性能
对双组分CeO2-WO3样品中脱硝效率最好的DCe(20)-W样品进行SCR抗水抗硫性能检测,结果如图3。控制温度为250 ℃,在30 min后通入0.02%SO2,发现DCe(20)-W样品脱硝效率从100%不断下降,在120 min后下降到68.91%,证明通入SO2对催化剂脱硝性能有一定的影响。之后继续通气催化剂效率没有明显的变化,这表明DCe(20)-W样品的抗硫性能较差。在360 min后停止通入SO2,DCe(20)-W样品没有表现出明显的效率回升,这可能是因为在铈基复合氧化物催化剂表面形成的硫酸盐物种是稳定的,堵塞了活性位点,并且这种反应是不可逆的[16]。相同条件下进行抗水实验,30 min时引入5%H2O,DCe(20)-W样品脱硝效率持续下降,在90 min后下降到84.22%。之后继续通气催化剂效率没有明显的变化,并且360 min停止通气后脱硝效率明显回升,这表明DCe(20)-W样品抗水性能良好。

图 3 250 ℃下双组分DCe(20)-W催化剂抗水抗硫性能Figure 3 Water and sulfur resistance of two-component DCe(20)-W catalyst at 250℃
2.2 催化剂物化性质分析
2.2.1 催化剂表面形貌及元素分析
为了探究样品的表面形貌和元素分布差异,对催化剂进行了SEM和EDS检测,结果如图4所示。单组分WO3催化剂(W-DS,图4(a))显示了WO3纳米棒的聚集性生长,呈现出海胆状。沉积CeO2后(DCe(20)-W,图4(b))可以明显观察到由纳米棒组装的珊瑚状结构,相比W-DS样品的形貌更加致密且纳米棒直径增加,这主要是因为CeO2晶粒沉积于WO3纳米棒上。这种粗糙的表面能够增加活性组分与反应气体组分之间的作用时间,有效改善异相催化反应的动力学,利于脱硝反应进行[31]。对应的EDS检测结果表明,与单组分催化剂(图4(c))表面只有W、O等主要元素相比,双组分催化剂(图4(d))表面还有明显的Ce元素出现,证明WO3以及CeO2在催化剂表面的成功负载。

图 4 单组分及双组分催化剂的扫描电镜照片Figure 4 Scanning electron micrographs of single-component and two-component catalysts
2.2.2 催化剂物相的XRD谱图分析
图5显示了催化剂的XRD谱图。单组分及双组分样品均在35°、38°、40°、53°、62°、70°、74°、76°、77°、83°、87°检测到PDF编号为44-1294的α-Ti晶相,该晶相对应于钛网基底。此外两种催化剂在14°、23°、28°、34°、36°、49°、55°、57°均出现PDF编号为85-2460的WO3峰,对应WO3的六方晶相,证实了WO3的成功沉积,与2.2.1节的EDS结果一致。但EDS中检测出的CeO2并没有XRD的对应峰,说明其在催化剂表面分散均匀,且多为非晶态,结晶度低,这有利于催化性能的提高[32]。

图 5 单组分及双组分催化剂的XRD谱图Figure 5 XRD patterns of single-component and twocomponent catalysts
2.2.3 催化剂的XPS分析
为了更好地分析催化剂表面元素价态的变化,对催化剂样品进行了X射线光电子能谱分析。在图6(a) O 1s的光谱中,通过进行峰拟合识别出两种表面氧物种,分别是530.52、530.79 eV处的晶格氧(表示为Oβ)和532.60、532.37 eV处的化学吸附表面氧(表示为Oα)[23]。催化剂对应元素比例列于表1中。由表1可知,DCe(20)-W中Oα/(Oα+Oβ)的比例(21.83%)高于W-DS(10.24%)。Oα是氧化还原反应中最活跃的氧,与Oβ相比具有更高的迁移性,因此活性更高,在SCR反应中能够促进NO氧化为NO2,实现“快速SCR”反应[33]。DCe(20)-W中Oα占比较高的原因可能是Ce4+与Ce3+之间进行能够电子转移,使得催化剂表面电荷不平衡,造成化学键不饱和,增加化学吸附氧的含量[34]。
由图6(b)可知,W6+物种在37.88、38.12 eV的W 4f5/2的峰和35.78、35.96 eV处的W 4f7/2峰,这表明催化剂表面形成了WO3物种[32]。DCe(20)-W的W 4f结合能低于W-DS样品,其可能原因为沉积CeO2使得W与Ce之间的相互作用增强,两物种间的电子转移改变了W物种周围的电子环境,会导致前者的W物种具有更高的电子云密度,降低结合能[15]。
图6(c)分析了两样品的Ce 3d光谱,对应u1和v1的峰是Ce3+的3d104f1电子态的主要代表,而v、v2、v3、u、u2和u3的峰是Ce4+所对应的3d104f0电子态[21]。Ce3+/(Ce3++Ce4+)的比值列于表1中,DCe(20)-W样品中的Ce3+比例为31.72%,这表明Ce和W物种的相互作用产生了更多的Ce3+[35]。Ce3+的存在会导致样品表面电荷不平衡和化学键不饱和,使得表面化学吸附氧提高,与O谱分析结果对应。沉积的CeO2与WO3相互作用增强,引入CeO2形成了更多的化学吸附氧Oα,可能会对脱硝反应起到促进作用[36]。
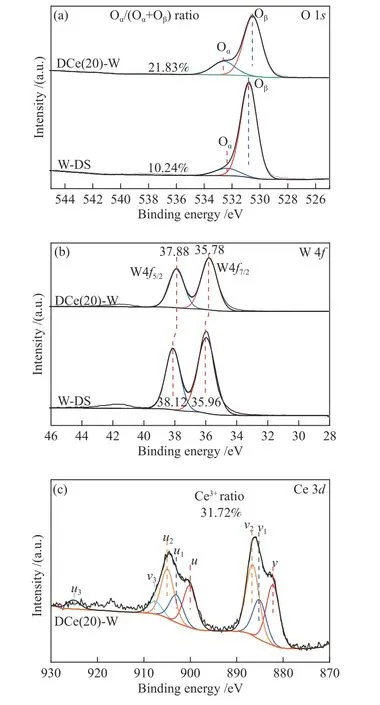
图 6 单组分及双组分催化剂的XPS谱图Figure 6 XPS patterns of single-component and two-component catalysts
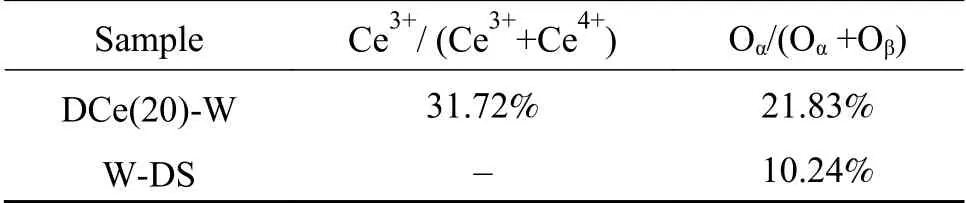
表 1 单组分及双组分催化剂表面原子浓度Table 1 Surface atomic concentration of catalysts
2.2.4 H2-TPR、NH3-TPD分析
根据已有研究发现,影响催化剂脱硝性能的主要因素包括氧化还原能力以及表面酸性,而氧化还原能力又是影响低温段催化剂脱硝性能的关键[18,37]。因此,本研究采用H2-TPR来进一步确定所制备催化剂的氧化还原能力,结果如图7(a)所示。DCe(20)-W在582 ℃的峰以及W-DS样品571 ℃的峰对应WO3的还原峰,表示WO3在表面上被还原[18,32]。同时,DCe(20)-W样品在516 ℃的峰对应表面氧的还原(Ce4+→Ce3+)[38,39],W-DS样品513 ℃的峰对应的是W-O-W桥接氧的还原峰[30]。然而,对这两种样品的总H2消耗量进行了定量检测,发现单组分W-DS样品为5.083 mmol/g,双组分DCe(20)-W样品为4.880 mmol/g,沉积CeO2导致H2消耗量有一定下降,这可能与催化剂表面引入的Ce3+以及CeO2-WO3对载体的覆盖有关[10]。结合上述分析可以看出,沉积CeO2后氧化还原能力略微下降,影响了样品脱硝性能的提升。脱硝数据表明,DCe(20)-W样品在200 ℃以上时脱硝效率明显提升,但对应氧化还原能力并没有明显改变,推断表面酸性的增强对中高温段脱硝反应起到了更重要的作用。为此,本研究对样品进行了NH3-TPD检测。
为分析沉积CeO2对表面酸性的影响机制,利用NH3-TPD实验检测单组分及双组分样品表面酸位点的数量和强度,结果如图7(b)所示。双组分DCe(20)-W样品在122 ℃附近的峰归属于弱吸收NH3,而在231、264以及374 ℃的峰归属于中强吸收NH3,在585 ℃的峰归属于强吸收NH3[38]。而单组分W-DS样品在189、310和368 ℃的峰归属于弱吸收及中强吸收NH3。对催化剂NH3的总吸附量进行检测,单组分WO3催化剂NH3总吸附量为0.652 mmol/g,而掺杂CeO2后催化剂表面的酸量显著增加,达到0.743 mmol/g。结合脱硝性能分析,总体酸性提高对200-350 ℃脱硝性能的改善有很大影响。从H2-TPR分析中可知,DCe(20)-W氧化还原能力对其脱硝性能影响不大,因此,其中高温段脱硝性能提升主要与酸性增强有关。
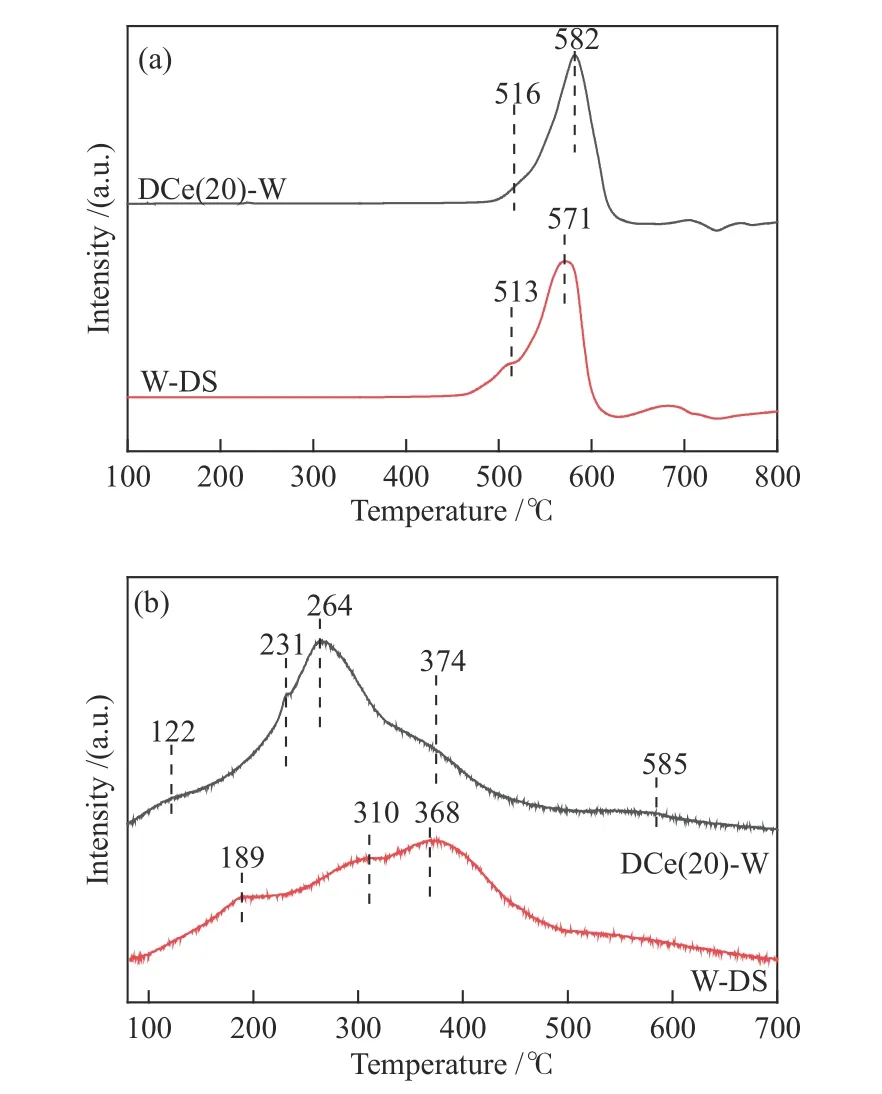
图 7 单组分及双组分催化剂的(a) H2-TPR,(b) NH3-TPD谱图Figure 7 Analysis of single-component and two-component catalysts(a): H2-TPR; (b): NH3-TPD
结合Liang等[3]的研究以及本研究NH3-TPD检测发现,单组分及双组分催化剂表面均具有Lewis酸以及Brønsted酸位点。此外Liu等[10]的研究指出CeO2主要具有弱酸和中强酸位,而WO3上存在中强酸位及强酸位。本研究中双组分催化剂掺杂CeO2后酸位点的分布有向低温方向移动的趋势且存在峰值强度的增强,这可能是因为CeO2引入了中强酸位点和弱酸位点,形成了WO3与CeO2的协同作用,产生更多的酸位点[15]。结合双组分催化剂中温段(250-350 ℃)的脱硝活性表现,证明了酸性增强有利于金属氧化物在中高温段脱硝性能的提高。
2.3 In-situ DRIFTS光谱分析
采用in-situDRIFTS光谱分析对DCe(20)-W样品的脱硝反应路径进行表征。
2.3.1 NH3吸附
DCe(20)-W样品在250 ℃下吸附NH3的in-situDRIFTS光谱如图8所示。在图8中,样品在976 cm-1波段可能是对NH3的弱吸附[40],在1034和1122 cm-1的谱带可能与Lewis酸位点上的配位NH3[41]有关,1410 cm-1处的谱峰可认为是与Brønsted酸位点结合的 N
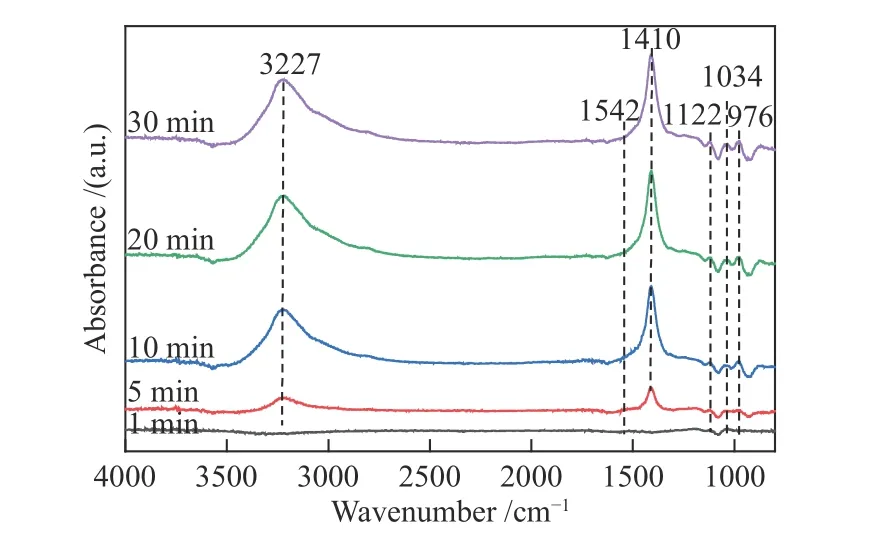
图 8 250 ℃下DCe(20)-W催化剂吸附NH3的in-situ DRIFTS光谱谱图Figure 8 In-situ DRIFTS spectral analysis of NH3 adsorption on DCe(20)-W catalyst at 250 ℃
[37],1542 cm-1的峰则可归于中间产物N物种[6],3227 cm-1的谱带为Lewis酸位点上吸附的共价态NH3[42]。所有波峰均在通气5 min后开始出现,并在10 min后相对稳定。随着吸附时间的延长,催化剂表面Lewis酸位点上的共价态NH3、Brønsted酸位点上的 N对应峰值均不断增大,表明吸附过程中Lewis酸与Brønsted酸位点均发挥作用,证实了Brønsted酸与Lewis酸位点对NH3的共同吸附。
2.3.2 NO+O2吸附
DCe(20)-W样品在250 ℃下吸附NO + O2的in-situDRIFTS光谱如图9所示。在图9中,样品各波段分别对应单齿硝酸盐(1196、1038、1122 cm-1)[37]、双齿硝酸盐(1559 cm-1)[6]、弱吸附的气态NO(917 cm-1),并且在通气1 min后就已出现并趋于稳定。而物理吸附NO2分子(1634、1653 cm-1)[43]在通气5 min后出现并始终保持较低峰值,表明NO较难被氧化为NO2。这与前述H2-TPR结论一致,即氧化还原性能降低,同时表明氧化还原能力的改变在中高温段脱硝反应中并非关键性因素。根据峰值强度分析,单齿硝酸盐峰值强度高,表明硝酸盐类物质容易在催化剂表面形成,并且较稳定。
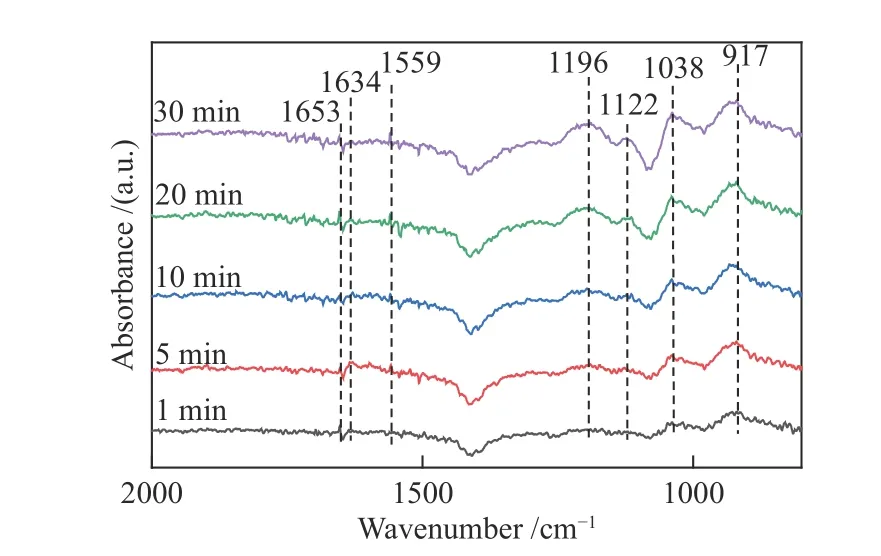
图 9 250 ℃下DCe(20)-W催化剂吸附NO+O2的in-situ DRIFTS光谱谱图Figure 9 In-situ DRIFTS spectral analysis of NO+O2 adsorption on DCe(20)-W catalyst at 250 ℃
2.3.3 吸附态NH3与NO+O2表面反应
DCe(20)-W样品在250 ℃下预吸附NH3然后转换为NO + O2的in-situDRIFTS光谱如图10所示。气体转换为NO + O2后,Lewis酸位点上吸附的共价态NH3(3217 cm-1)、与Brønsted酸位点结合的 N(1409 cm-1)、弱吸附态NH3(975 cm-1)吸收峰均发生变化。随时间的延长,峰强度逐渐减弱并在通气10 min后消失,这表明吸附在Lewis酸以及Brønsted酸位点上的NH3物种会与气态NO发生反应,这种反应会导致NH3物种的消耗。1194 cm-1波段出现的新峰对应单齿硝酸盐,但Lewis酸上的配位NH3(1038、1120 cm-1)对应峰没有明显变化。研究表明[44],吸附态的NH3只有通过进一步活化才能参与后续反应。据此,以上结果证实DCe(20)-W催化剂遵循Eley-Rideal(E-R)反应机制[37],即中温段(250 ℃)两种吸附态NH3很容易直接与气态NOx反应。

图 10 250 ℃下DCe(20)-W催化剂预吸附NH3 30 min后吸附NO + O2的in-situ DRIFTS光谱谱图Figure 10 In-situ DRIFTS spectral analysis of DCe(20)-W catalysts pre-adsorbed by NH3 for 30 min and reacted with NO + O2 at 250 ℃
综合上述分析,得到DCe(20)-W催化剂的反应路径: NH3物种首先以 N离子和配位NH3的形式分别吸附在Lewis酸和Brønsted酸的表面。随着温度升高,催化剂对NH3吸附能力减弱,但脱氢活化能力增强。在中温段(250℃)O2存在的条件下,吸附态的NH3转变为中间产物酰胺类N-参与反应。具体反应路径如下(*表示表面活性位点):

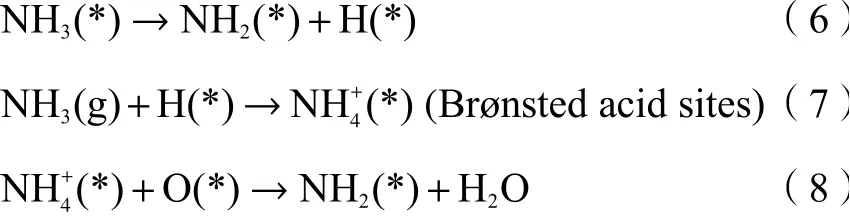
随后中间产物直接与气态NO反应生成NH2NO,该产物高温易分解,并最终转化为N2和H2O。
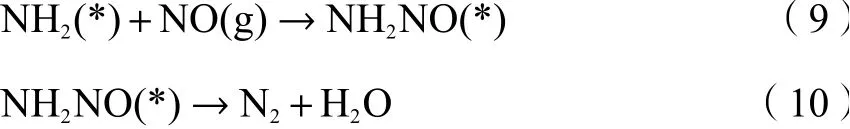
3 结 论
本研究采用电沉积加水热法在钛网基底上合成了WO3纳米棒,然后采用电沉积法成功负载了CeO2,作为脱硝催化剂,并探索脱硝反应机理。通过检测分析得到结论如下。
电沉积CeO2时长20min为双组分样品最佳负载条件,该条件制备的样品在250-350 ℃脱硝效率达到100%,活性窗口扩大至200-400 ℃,符合实况下柴油车尾气装置正常工作温度范围。
CeO2掺杂于WO3表面后引入了Ce3+,增加了化学吸附氧Oα的比例,但催化剂的氧化还原性能未呈现明显提高。由于中高温段脱硝反应性能更多依赖与表面酸性位点,CeO2的负载有效引入了弱酸和中强酸位点,导致催化剂表面酸性增加,促进了中温段(250-350 ℃)脱硝性能的提高。
In-situDRIFTS结果显示DCe(20)-W样品遵循(E-R)反应机制,两种吸附态NH3均可以迅速与气态NOx反应。酸性位点为脱硝反应主导因素,NH3物种首先以 N离子和配位NH3的形式分别吸附在Lewis酸和Brønsted酸的表面,进一步反应生成中间产物酰胺类 N并最终在高温下分解为N2和H2O。
