Co2+调控WOx表面Brönsted酸和氧空位含量用于提高1-己烯环氧化性能
2022-12-14邢向英王会香王连成吕宝亮
邢向英 ,王会香 ,王连成 ,吕宝亮
(1. 中国科学院山西煤炭化学研究所 煤转化国家重点实验室, 山西 太原 03001;2. 中国科学院大学, 北京 100049)
烯烃环氧化物是一类重要的有机合成中间体,因环氧三元环结构在电荷诱导作用下极易开环,并可以快速与含有活泼氢的基团,如胺基、羧基、羟基和酚羟基等生成高附加值化学品,从而被广泛应用于石油化工、精细化工、医药合成、农业以及建筑等领域[1-4]。铁基费托合成油品中含有大量直链烃类(> 95%),其中,α-烯烃比例占50%以上[5-8]。然而,碳数相近的长链α-烯烃因物理性质极相似,导致其分离成本增大,因此,这些烯烃混合物被直接加氢饱和制成附加值有限的油品,从而造成了大量优质长链α-烯烃资源的浪费[9]。通过环氧化方法将长链α-烯烃转化为长链1,2-环氧烷烃,官能团的转化不仅能增大各物质间物理性质差异,便于产物的分离,同时可提高长链α-烯烃的附加值,增加铁基费托合成油的经济效益。此外,这一做法也符合化石能源清洁高效利用,以及推动其下游产品高附加值综合利用的新战略发展目标[10]。
常用的烯烃环氧化方法有卤醇法、有机过酸法、空气/氧气氧化法和H2O2氧化法[11,12]。然而,卤醇法和有机过酸法不仅过程复杂、副产物多,更大的弊端是会造成环境污染。虽然空气/氧气氧化法是一种环境友好型的环氧化方式,但因氧气分子活化需要较高温度,且高温下反应产物较复杂,因此,其实际生产受高温、高压限制[13]。相比之下,H2O2成本低、易活化、绿色无污染,因此,被广泛应用于烯烃的环氧化研究中[14-16]。然而,在没有催化剂的作用下,低含量H2O2(<50%)无法直接将烯烃环氧化;若使用高含量的H2O2又存在易燃易爆的安全隐患[11]。因此,从经济和环境友好角度出发,设计以低含量H2O2为氧化剂,应用于长链α-烯烃环氧化的高效催化剂具有广阔的前景。
通过文献调研发现,目前以H2O2为氧源的环氧化催化剂主要有钛硅分子筛、多金属氧酸盐和甲基三氧化铼等[16-18]。钛硅分子筛由于孔道尺寸的限制,其对长链α-烯烃的环氧化效果不理想;多金属氧酸盐等均相催化剂虽然可以高效环氧化长链α-烯烃,但在反应结束后很难分离,且极易发生活性组分的流失。相比之下,过渡金属氧化物价态丰富、储量大、结构多样且易分离回收,因此是比较理想的长链α-烯烃环氧化催化剂。
WO3作为一种过渡金属氧化物固体酸催化剂,其活性高、化学性质稳定,具有可调的带隙结构,已广泛应用于电池、传感以及催化等众多领域[19-22]。目前,WO3或带有氧缺陷的WOx已经被应用于H2O2体系下的烯烃催化环氧化反应。例如,Ghosh等[23]制备了Ag/WO3纳米棒,发现其在以H2O2为氧源的苯乙烯环氧化体系中表现出较高活性;Zhang等[24]通过高温热解制备的H2OWOx@C(ZIF)催化剂在H2O2的作用可将环辛烯有效转化为环氧辛烷;Maheswari等[25]制备了WOx/SBA-16催化剂,其可催化环辛烯环氧化,而Gao等[26]合成的WOx-MCF催化剂可将环1,5-二烯转化为相应的环氧化物。研究发现,当H2O2为氧化剂时,其和催化剂作用形成的M-O-OH是烯烃环氧化的活性中间体,而这一中间体的形成与金属表面的氧缺陷密不可分[14,24,27],且H2O2的利用率可以根据环氧化合物的生成量来计算[28]。此外,环氧化产物的分布和WO3表面的Brönsted酸(B酸)含量密切相关,B酸含量增多会促进环氧化合物的水解,以致环氧烷烃的选择性下降[29]。因此,用科学有效的手段在增加WO3表面氧缺陷的同时减少其表面B酸含量,将是提高WO3活性的有效途径之一。
金属阳离子掺杂是一种有效的化学改性方法,一方面,掺杂到WO3基体中的阳离子因具有不同的离子态和配位结构,导致基体中八面体结构发生畸变,可以产生较多的氧空位[30-33];另一方面,金属阳离子难免和WO3表面酸性羟基反应从而减少WO3表面B酸位点数量。课题组前期研究发现,Ni2+可以有效降低MoO3表面B酸含量,同时增加表面氧空位含量,从而提高了其氧化脱硫性能[34];W与Mo为同族元素,因此,理论上阳离子改性对WO3表面B酸应具有相似的结果。钴(Co)是重要的过渡金属之一,其离子半径与W离子相当,被认为是WO3的优良掺杂剂[35]。比如,Abbas等[36]报道Co掺杂到金属氧化物纳米颗粒中,由于氧空位等结构缺陷,提高了颗粒的抗癌活性。基于以上研究和分析,笔者计划引入Co2+对WO3进行掺杂来改变WO3表面B酸和氧空位含量,并进一步探究其长链α-烯烃环氧化性能。
为了均匀制备本身就带有氧缺陷的高活性催化剂,以WCl6为钨源,乙醇为溶剂,采用动态法合成了WOx,并在过程中直接引入Co(NO3)2·6H2O作为Co2+源成功制备了Co-WOx催化剂。以H2O2为氧化剂,1-己烯环氧化为探针反应,系统测试了Co2+引入前后WOx催化剂环氧化性能的变化以及反应参数对环氧化的影响规律,并结合催化剂结构表征结果阐述了内在原因,最后对环氧化机理做了深入讨论。
1 实验部分
1.1 实验试剂
氯化钨(WCl6,99%)、1-己烯(C6H12,99%)和苯甲醚(C7H8O,99%)购于阿拉丁;乙腈(CH3CN,99%)、硝酸钴(Co(NO3)2·6H2O,99%)和30% H2O2均购买于国药集团化学试剂有限公司;无水乙醇(EtOH,99%)购于天津市北辰方正试剂厂。所有实验试剂均为分析纯,实验用水为电阻率为18.21 MΩ/cm的蒸馏水。
1.2 催化剂的制备
WOx催化剂的制备:2 mmol WCl6溶于50 mL无水乙醇中,超声至WCl6完全溶解。将上述溶液转入100 mL的聚四氟乙烯反应釜中密封,然后固定在均相反应器(KLJX-12,烟台高新区科立自控设备研究所)内160 ℃反应12 h,旋转频率30 Hz。反应产物分别经三次水和乙醇洗涤,随后置于60 ℃烘箱内干燥后即得纯WOx。
Co-WOx催化剂的制备:采用直接引入法,2 mmol WCl6完全超声溶解后,分别向溶液中加入0.4、0.2和0.1 mmol Co(NO3)2·6H2O,待其全部溶解后转入100 mL反应釜中,后经相同制备过程得到Co-WOx-x样品(x= 0.2、0.1和0.05,为Co/W物质的量比)。
1.3 催化剂的表征
文中催化剂的晶型结构由X射线粉末衍射仪(XRD,D8 Advance)得到,测试过程使用单色CuKα靶(波长为1.5406 Å),工作电压和电流分别为40 kV、40 mA,扫描速率为2(°)/min,扫描5°-90°。样品形貌初步通过热场发射扫描电镜(SEM,JSM-7900F)观察,工作电压10 kV、电流0.1 nA。热场发射透射电子显微镜(TEM,JEM-2100F)用来进一步观察催化剂的形貌结构和高分辨图中晶格条纹间距的变化,工作电压200 kV、电流200 μA,晶格条纹间距使用Digital Micrograph软件量取,取多组数据的平均值。X射线光电子能谱(XPS,AXIS ULTPA DLD)提供催化剂的表面原子化学价态信息,测试使用单色AlKα靶源,以污染碳的C 1s峰作为定标标准。样品的拉曼光谱(Raman,LabRAM HR Evolution)使用可见光源波长为514 nm,谱图采集前进行硅片校正。催化剂表面酸含量的变化采用原位氨气红外光谱(NH3-FTIR,Bruker 80 V)测定。
1.4 催化剂的环氧化性能测试
以30% H2O2为氧化剂,1-己烯环氧化为探针反应,测试所得样品催化反应活性。反应过程如下:将2 mmol 1-己烯、2 mmol H2O2(30%)、3.5 g乙腈和0.05 g催化剂加入体积为15 mL的微型反应釜内,超声分散2 min后将反应釜拧紧,并将其固定在均相反应器内,在60 ℃下以30 Hz的转速进行旋转反应。反应完成后,向溶液中加入0.17 g苯甲醚作为内标,混合均匀离心取上层清液,通过GC-MS对产物进行定性分析,用配备有FFAP色谱柱(50 m×0.32 mm×0.32 μm)的GC-920气相色谱对产物进行定量,利用标准曲线法计算1-己烯的转化率和1,2-环氧己烷的选择性。实验分别探究了Co/W物质的量比、催化剂用量、H2O2用量、反应时间、反应温度对1-己烯环氧化反应的影响。1-己烯转化率和产物选择性,H2O2的利用率和转化率的计算公式如下:

式中,n0是1-己烯或者H2O2初始物质的量,nt是反应t时间后剩余的物质的量;ni是环氧化反应产物物质的量。H2O2利用率以生成的1,2-环氧己烷为参考[28];H2O2转化率采用铈量法[37],以0.05 mol/L的硫酸铈为滴定剂,邻二氮菲亚铁盐为指示剂。
2 结果与讨论
2.1 催化剂的表征
首先对不同Co/W物质的量比的样品Co-WOxx进行XRD表征,结果如图1(a)所示。可以看出,催化剂的结晶度均较差,纯WOx样品中位于23.27°和47.57°的强衍射峰归属于单斜相WO2.72(JCPDS#36-0101)的(010)和(020)晶面[38],可见样品具有明显的择优生长取向,其(010)晶面立体结构如图1(b)。由于(010)晶面上氧原子浓度较大,在(100)和(001)面原子模型中更易体现,因此,WOx(010)面更易结合钨源沿[010]方向生长。从图中可以看出,这两个衍射峰在引入Co2+后没有发生明显的偏移,因此,Co2+没有影响WOx晶型结构和主生长方向。此外,XRD谱图中也没有出现Co物种的衍射峰,主要是因为Co含量本身少,且部分进入晶格或含Co物质结晶度较差的原因。
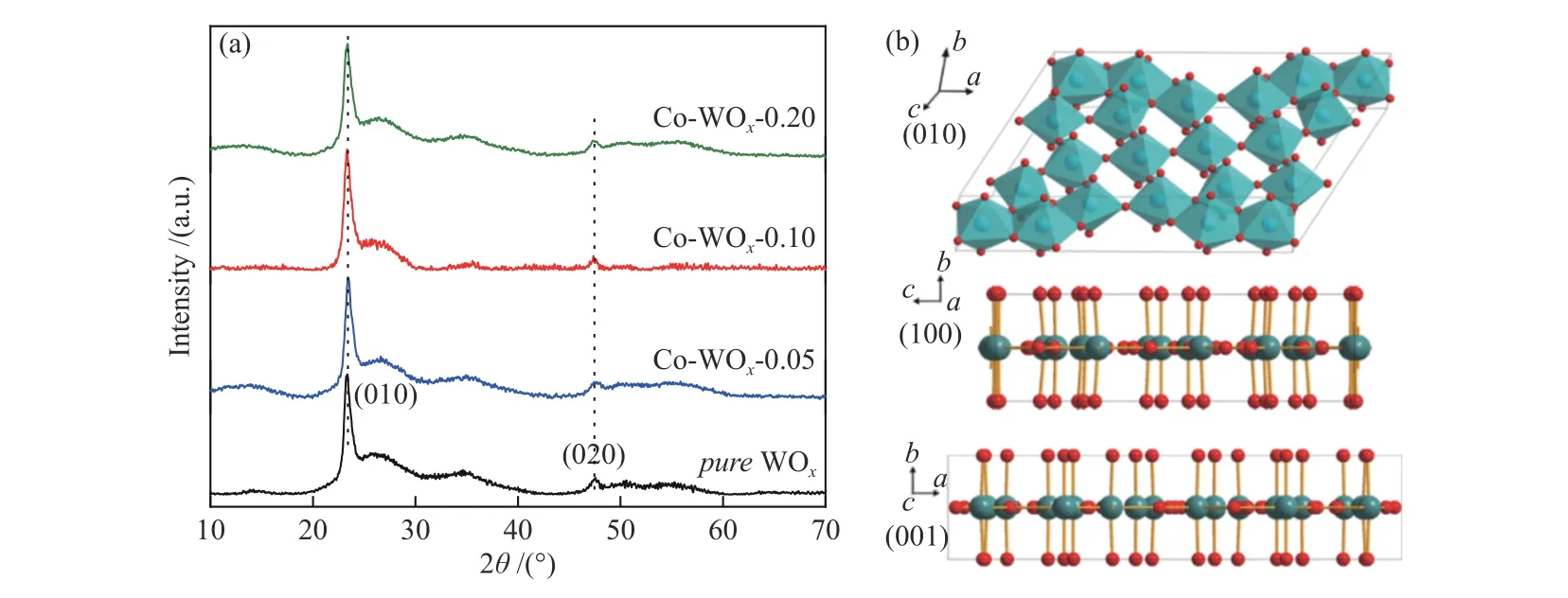
图 1 (a)纯WOx和Co-WOx-x样品的XRD谱图;(b)(010)晶面的立体图及(100)和(001)晶面的原子排布Figure 1 (a) XRD patterns of pure WOx and Co-WOx-x samples, (b) Three-dimensional view of the (010) crystal plane,and the atomic arrangement of the (100) and (001) crystal planes
催化剂的SEM形貌如图2所示。可以看出,纯WOx为纳米线(直径约10 nm)组成的蓬松状纳米球,直径约为800 nm,团聚较严重。当x= 0.05和0.1时,样品Co-WOx-0.05和Co-WOx-0.1结构没有改变,但是分散性有所改善,可能是Co2+的引入略微减缓了WOx的结晶速率,从而使样品得以分散生长;Co-WOx-0.1的纳米线长度有所减小,同样说明Co2+在一定程度上有阻碍WOx生长的作用。当x =0.2时,Co-WOx-0.2的纳米线长度进一步减小,纳米球之间团聚较纯WOx更为严重,而且出现了块状结构,因为过量的Co2+阻碍了WOx纳米线的生长,从而使得球状中心过多,导致相互团聚。由此可见,Co2+含量过高不利于WOx形貌结构的维持。为了进一步分析催化剂的微观结构差异,对纯WOx和催化性能较好的Co-WOx-0.1样品(后期催化实验证实)进行了更详细的结构分析。

图 2 纯WOx和Co-WOx-x样品的SEM照片Figure 2 SEM images of different samples: ((a), (b)) pure WOx; Co-WOx-x: ((c), (d)) x=0.05; ((e), (f)) x=0.1; ((g), (h)) x=0.2
从图3(a)、3(b)和图3(d)、3(e)的TEM照片中可明显看到,相比于纯WOx,Co-WOx-0.1纳米球上的纳米线长度减小。图3(c)是纯WOx的HRTEM照片,0.37 nm的晶格条纹间距对应(010)晶面[38],说明纳米线沿[010]方向生长,对应于XRD中的择优生长取向。Co-WOx-0.1催化剂的(010)晶格条纹间距为0.38 nm(图3(f)),变化不大,但可以看出其非晶层的厚度增加。原因主要有两个方面:一是,体系中的Co2+作用于WOx表面,影响了WO3的进一步结晶;二是,在合成过程中部分Co2+进入WOx的晶格产生原子替代作用,导致W6+配位不饱和,使其有序性和结晶度进一步减弱,同时极大可能伴随氧空位的增加。
XPS表征被用来进一步检测样品表面原子的价态和化学环境。图4(a)是两个样品的全谱,谱图可以看出,Co-WOx-0.1样品中除了W和O元素外出现了Co的峰,Co元素含量较低导致其信号不强。对Co 2p进行拟合后(图4(b)),在781.14和797.07 eV出现了能量差为15.93 eV的两个峰,分别归属于Co 2p3/2和Co 2p1/2,两者均对应于Co2+的特征峰[39]。如图4(c)所示,对纯WOx的W 4f峰进行拟合处理,34.44、37.31、35.56和37.55 eV分别对应于W5+4f7/2、W5+4f5/2和W6+4f7/2、W6+4f5/2结合能。在Co-WOx-0.1催化剂中,这四个峰的数值略微向高结合能偏移,分别为34.52、37.39、35.61和37.57 eV,由此可见,阳离子Co2+的引入微降低了W原子电子云密度。如图4(d)所示,位于530.14、531.29和532.70 eV的峰分别归属于纯WOx的晶格氧、氧空位和吸附H2O[35]。同样地,Co-WOx-0.1的O 1s峰也向高结合能方向移动,上述三种氧结合能分别位于530.28、531.38和532.81 eV。通过面积定量分析可知,Co-WOx-0.1中W5+和氧空位(Ov)含量(37.37%、34.30%)均高于纯WOx中两者含量(29.97%、25.20%),此结果证实Co2+的引入确实可以增加WOx氧空位。

图 3 ((a),(b))纯WOx和((d),(e))Co-WOx-0.1的TEM照片;(c)纯WOx和(f)Co-WOx-0.1的HRTEM照片Figure 3 TEM images of ((a), (b)) pure WOx and ((d), (e)) Co-WOx-0.1, HRTEM images of (c) pure WOx and (f) Co-WOx-0.1
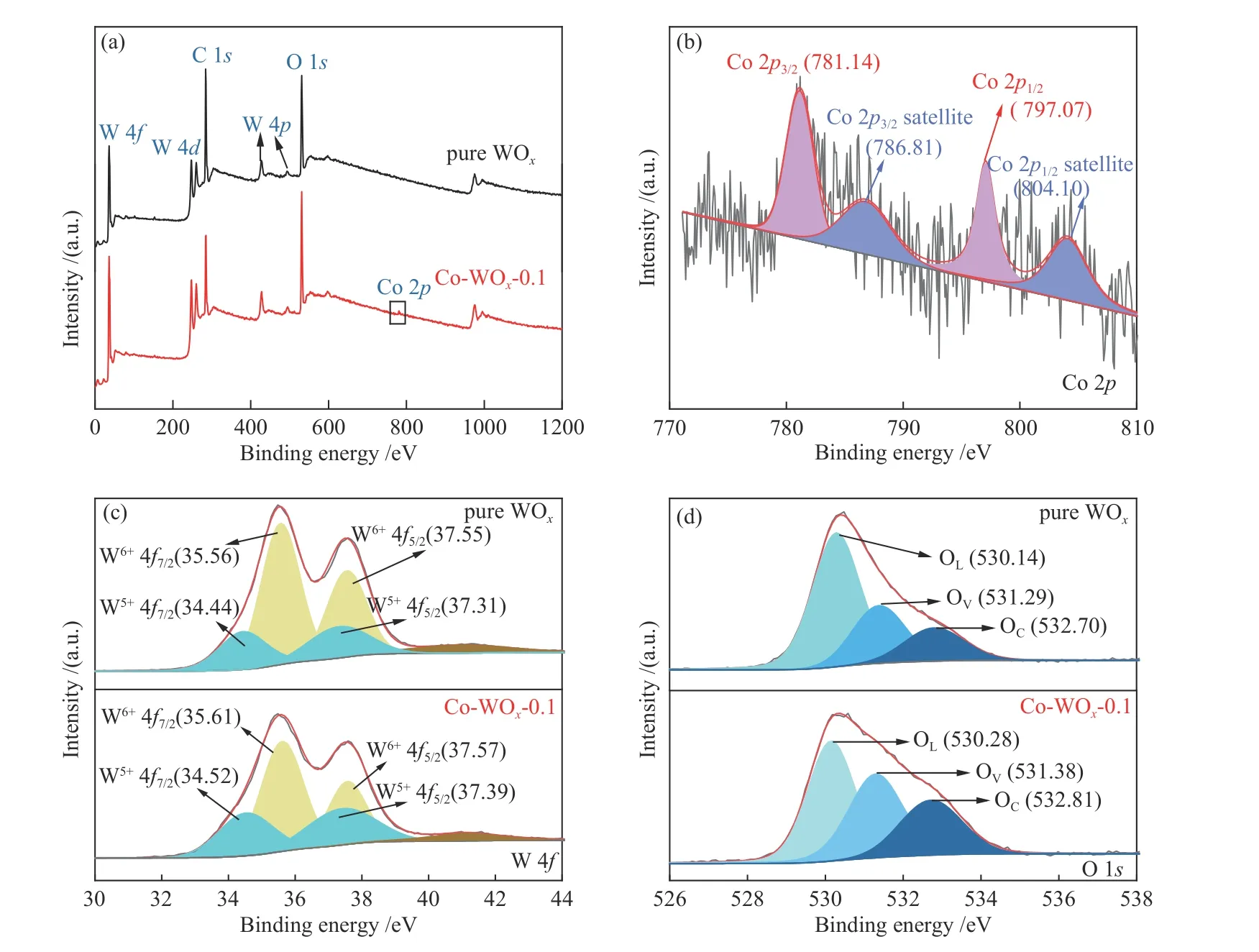
图 4 纯WOx和Co-WOx-0.1的XPS谱图Figure 4 XPS spectra of pure WOx and Co-WOx-0.1 samples: (a) full spectrum; (b) Co 2p; (c) W 4f and (d) O 1s
纯WOx和Co-WOx-0.1的Raman测试结果如图5所示,对纯WOx而言,698.3和810.4 cm-1的峰归属于O-W-O的伸缩振动,261.6和323.6 cm-1的峰归属于W-O-W的弯曲振动[35]。在Co-WOx-0.1样品中四个特征峰均出现,也没有出现Co物种的信号峰,说明催化剂WOx的结构没有大的变化,然而与纯WOx相比,有三个WOx峰位置出现了偏移,峰的相对强度也发生了变化,说明Co2+的引入还是对W周围的化学环境引起了变化[35]。
催化剂表面B酸含量对环氧化反应有着重要的影响,因此,对纯WOx和Co-WOx-0.1样品做原位NH3-FTIR来检测其表面酸性,结果如图6所示。一般地,NH3分子吸附在L酸(不饱和金属离子)位点时,氮的孤对电子配位到L酸部,δN-H吸收峰位置约在1610 cm-1;在B酸(表面酸性羟基)位点,NH3接受质子形成吸收峰位置大约为1430 cm-1[40]。实际情况中,因样品差异,测试温度、仪器程序升温等条件参数的不同会使峰的位置发生偏移。如图6(a)所示,纯WOx表面主要以B酸为主,而Co-WOx-0.1谱图(图6(b))中L酸含量增加比较明显。具体对1410、1621、1406和1628 cm-1处的峰进行积分,计算L酸峰面积与B酸峰面积比值(AL/AB)。如表1所示,Co2+掺杂后AL/AB明显提高,一方面,引入的Co2+和WOx表面羟基(B酸位点)进行连接生长作用,从而减少了B酸位点数量;另一方面,Co2+掺杂后L酸含量增加,主要是由Co2+掺杂后引起了氧空位数量增多导致的,Gonzalez等已经证实了L酸含量与氧空位数量呈正相关性[41]。
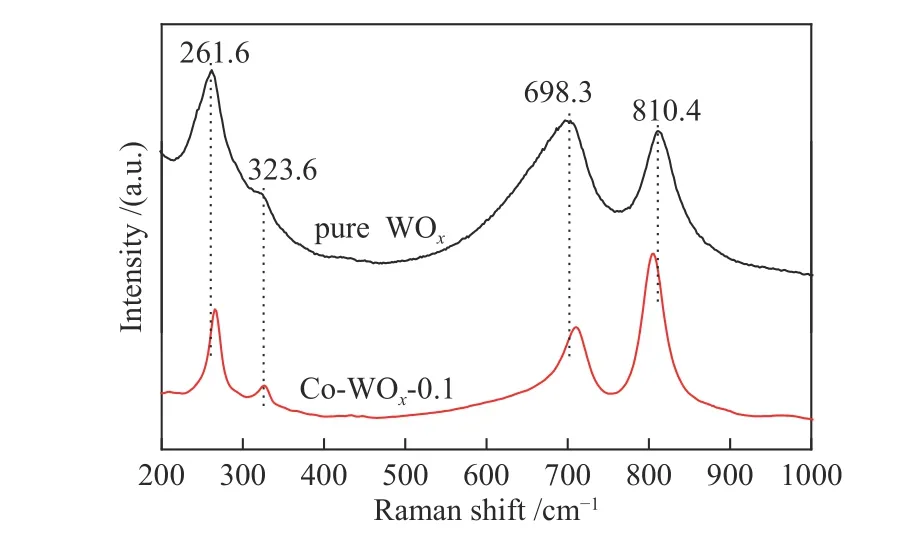
图 5 纯WOx和Co-WOx-0.1的Raman光谱谱图Figure 5 Raman spectra of pure WOx and Co-WOx-0.1samples
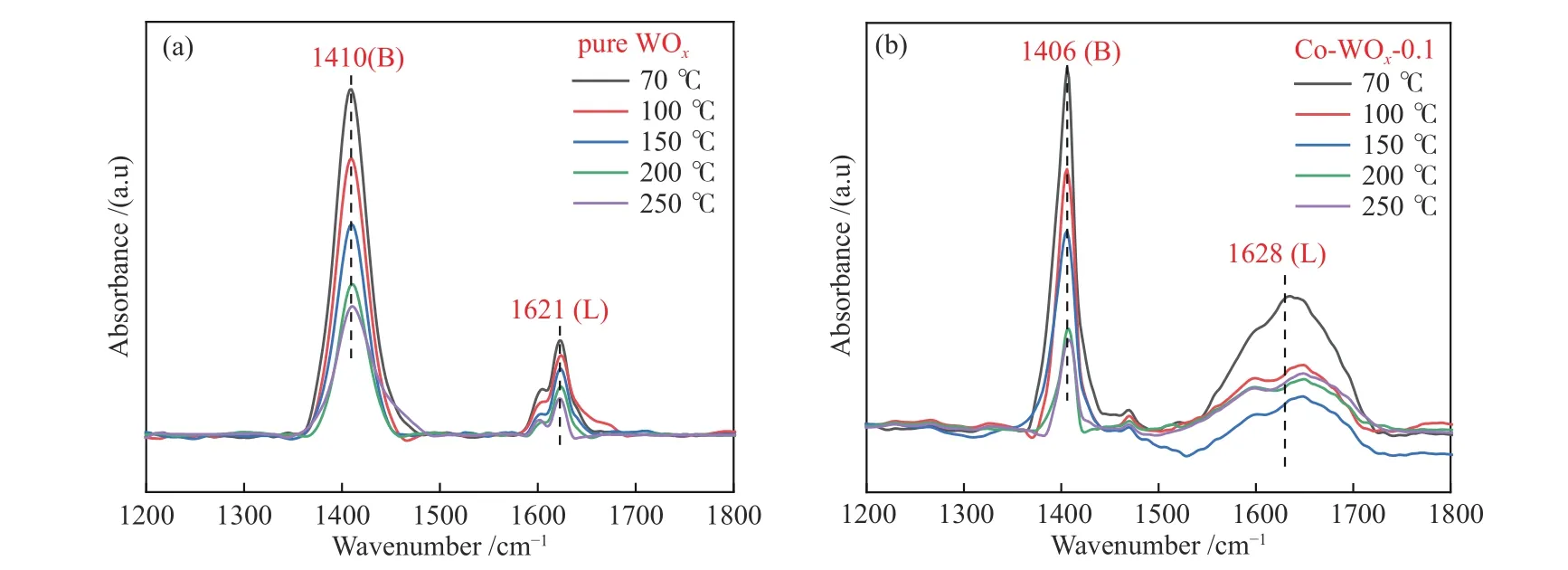
图 6 原位NH3-FTIR吸收光谱谱图Figure 6 In-situ NH3-FTIR absorption spectra: (a) pure WOx; (b) Co-WOx-0.1
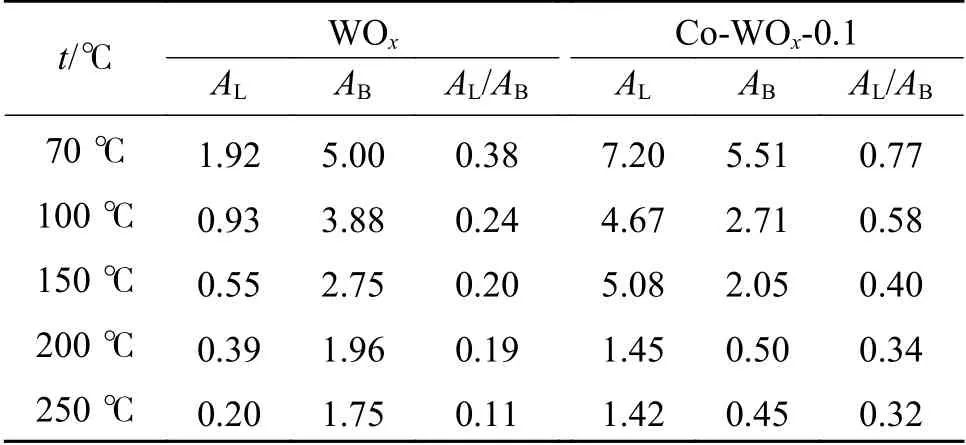
表 1 纯WOx和Co-WOx-0.1的原位NH3-FTIR峰面积Table 1 In-situ NH3-FTIR peak areas of pure WOx and Co-WOx-0.1
2.2 催化剂环氧化性能测试
首先利用GC-MS(QP-2010)对纯WOx环氧化1-己烯的产物进行定性分析(气化室和检测器温度分别为230和250 ℃,以10 ℃/min的升温速率从50 ℃升到230 ℃,保留时间20 min),产物主要有:正戊醛(n-valeraldehyde)、1,2-环氧己烷(1,2-epoxyhexane)、2-醇-1-己烯(2-alcohol-1-hexene)、2-羰基-1-己烯(2-carbonyl-1-hexene)、2-羰基-1-己醇(2-carbonyl-1-hexanol)、正戊酸(n-valeric acid)以及1,2-己二醇(1,2-hexanediol)。接着进行了两组空白实验:一是,在不加催化剂的情况下,该体系没有产物生成,说明30%的H2O2并不能将1-己烯直接环氧化;二是,仅加入催化剂WOx和底物1-己烯,在没有H2O2的条件下,也没有检测到反应产物。因此,1-己烯环氧化反应的进行是WOx与H2O2共同作用的结果。基于以上分析,下面具体讨论催化剂催化1-己烯环氧化反应中Co/W物质的量比、催化剂用量、反应时间、H2O2用量和反应温度五种因素对产物选择性和底物1-己烯转化率的影响。
2.2.1 Co/W物质的量比的影响
实验条件:2 mmol 1-己烯、2 mmol 30% H2O2、0.05 g催化剂,60 ℃,8 h,n(H2O2∶1-己烯) = 1。如图7(a)所示,纯WOx催化反应中,1,2-环氧己烷的选择性仅有26.9%,1-己烯的转化率为45.1%。其中,1,2-己二醇和2-羰基-1-己醇的选择性较高,主要原因是纯WOx表面B酸含量较高,体系内生成的1,2-环氧己烷在B酸的作用下进一步发生水解生成1,2-己二醇[29];一部分1,2-己二醇在氧化体系中被进一步氧化成2-羰基-1-己醇[26]。当Co/W物质的量比为0.1时,1,2-环氧己烷的选择性最高为55.7%,1-己烯的转化率为39.8%;目标产物选择性的提高与Co-WOx-0.1表面的B酸位数量降低有直接关系,其有效减少了环氧产物的水解;而底物转化率的降低也与表面B酸含量的减少有关,1,2-己二醇的生成本质上是连续反应[42],即底物首先转化成1,2-环氧己烷,1,2-环氧己烷在表面B酸的催化作用下水解。根据化学平衡原理,当水解反应被抑制后,进而底物生成环氧己烷的反应也被抑制,导致最终1-己烯的转化率略有降低。当n(Co/W) = 0.05,WOx表面B酸的水解作用依然比较严重,进而影响了1,2-环氧己烷的选择性。然而,当Co2+掺入量(n(Co/W) = 0.2)较高时,体系中的正戊醛选择性略有提高,1-己烯的转化率进一步降低,这可能是因为较多的Co2+会自生长形成孤立的CoOx物种,负载于WOx表面,占据了部分活性位点,且其具有断键、氧化能力,容易直接将部分1-己烯的双键断键氧化成正戊醛[43,44],因此,Co/W的最佳物质的量比为0.1。
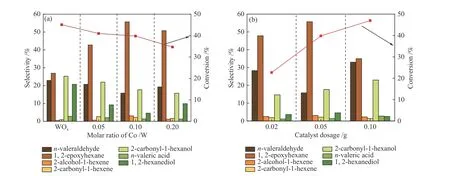
图 7 1-己烯环氧化反应的影响因素Figure 7 Influence factors of epoxidation toward 1-hexene: (a) molar ratio of Co/W; (b) catalyst dosage
2.2.2 催化剂用量的影响
实验条件:2 mmol 1-己烯,2 mmol 30% H2O2,Co-WOx-0.1,60 ℃,8 h,n(H2O2∶1-己烯) = 1。如图7(b)所示,当催化剂用量为0.02 g时,少量的催化剂不能提供充足的活性位点将H2O2活化,因此,体系内1.2-环氧己烷选择性和1-己烯的转化率均较低。然而,当催化剂用量增大到0.1 g时,1-己烯的转化率升高趋势有所减弱。这是因为催化剂用量增加时,其颗粒之间难免会团聚,从而影响反应物和产物的传质,吸附于催化剂表面的1-己烯脱附较难,从而被过度氧化生成正戊醛[34,45];同时,催化剂含量增多意味着单位体积内的B酸含量也会增多[25],部分1,2-环氧己烷在催化剂表面生成1,2-己二醇,1,2-己二醇也会在催化剂表面被进一步氧化成2-羰基-1-己醇。因此,催化剂最佳用量为0.05 g时体系的反应比较理想。
2.2.3 反应时间的影响
实验条件:2 mmol 1-己烯,2 mmol 30% H2O2,0.05 g Co-WOx-0.1,60 ℃,n(H2O2∶1-己烯) = 1。如图8(a)所示,从2 h到10 h,1-己烯的转化率随着时间的延长不断增加;1,2-环氧己烷选择性在2-8 h有着相同的增长趋势,8 h时达到最高。然而,在连续反应中[42],当反应时间低于8 h时,1,2-环氧己烷的生成速率大于其水解速率;随着反应时间的延长,体系中H2O2的含量降低,1,2-环氧己烷的生成速率小于1,2-环氧己烷水解生成1,2-己二醇的速率,此时导致体系中1,2-己二醇的含量增多,1,2-环氧己烷物质的量减少,选择性下降。综上,反应的最佳时间是8 h。
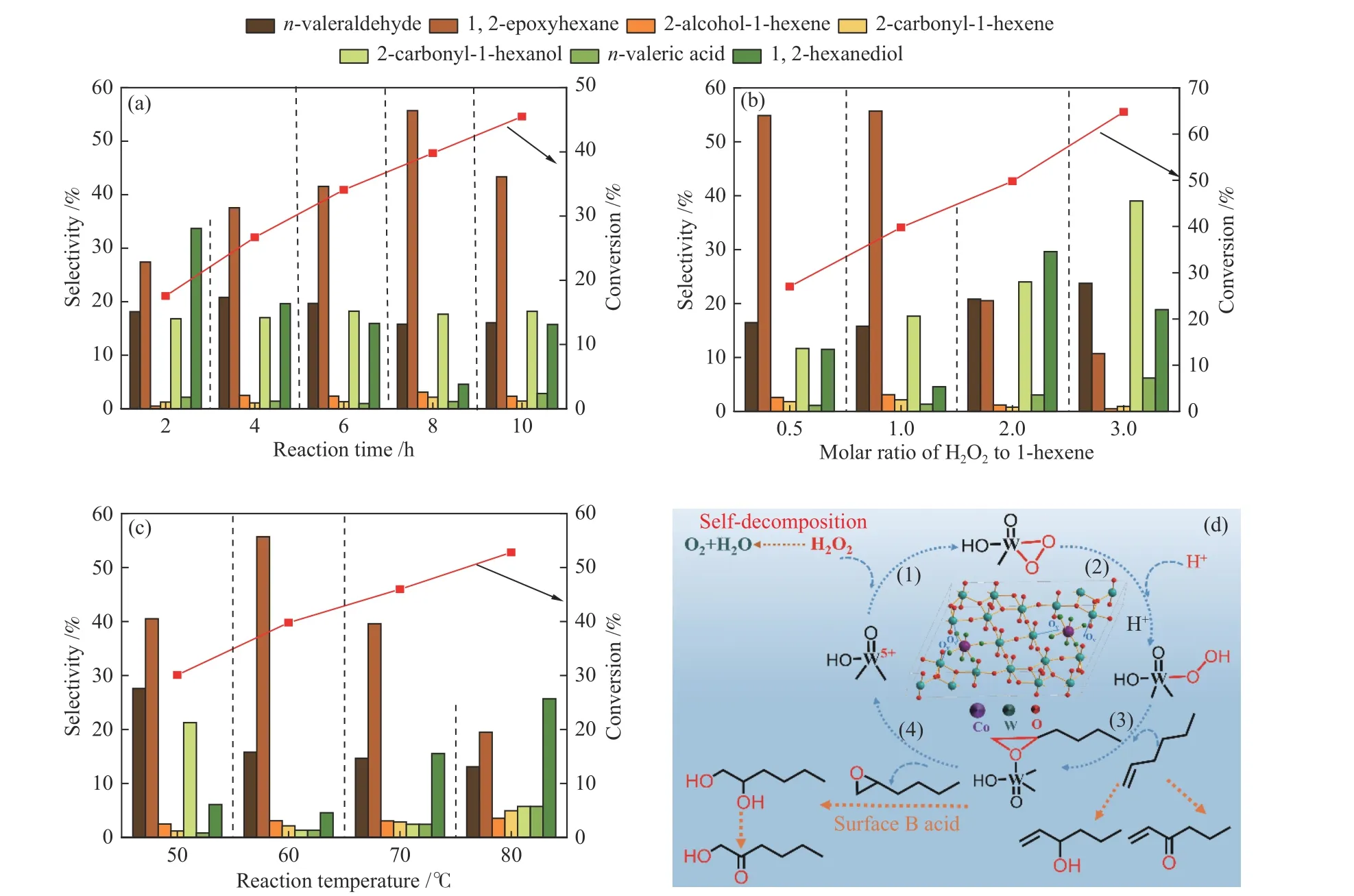
图 8 1-己烯环氧化反应的影响因素Figure 8 Influence factors of epoxidation toward 1-hexene: (a) reaction time; (b) molar ratio of H2O2 to 1-hexene;(c) reaction temperature; (d) possible epoxidation mechanism of 1-hexane
2.2.4 H2O2与1-己烯物质的量比影响
实验条件:2 mmol 1-己烯,2 mmol 30% H2O2,0.05 g Co-WOx-0.1,60 ℃,8 h。从图8(b)可以看出,环氧体系中H2O2与1-己烯的最佳物质的量比为1∶1。当n(H2O2∶1-己烯)<1时,1-己烯的转化率较低;然而当n(H2O2∶1-己烯)>1时,虽然1-己烯的转化率不断上升,但是1,2-环氧己烷的选择性大幅下降,原因主要有以下两点:一是,随着H2O2用量的增加,体系中的水也不断增加,在B酸的作用下,1,2-环氧己烷的水解反应更容易发生;二是,体系的氧化能力也随H2O2比例的提升而增强,使体系内正戊酸和2-羰基-1-己醇的选择性增大,从而导致1,2-环氧己烷的选择性降低。
H2O2与1-己烯的物质的量比不仅影响产物的选择性和底物的转化率,对H2O2自身转化率和利用率也有直接影响。如表2所示,0.05 g Co-WOx-0.1催化环氧化体系中,H2O2比例增加导致体系中有较多的副反应发生,易消耗H2O2,致使H2O2的转化率随着H2O2的比例增加而增加[37],但是当n(H2O2∶1-己烯)>1,H2O2的利用率随H2O2比例的提升而降低,主要是因为部分1,2-环氧己烷水解生成了1,2-己二醇,环氧化合物的物质的量降低影响了H2O2的利用率。当n(H2O2∶1-己烯)=1,Co-WOx-0.1催化反应中的H2O2转化率比其在纯WOx催化反应中的高,这可能与Co-WOx-0.1表面氧空位的增加有关,氧空位增加利于活化H2O2形成W-O-OH,增加了H2O2的转化率[14,24]。

表 2 H2O2的利用率和转化率Table 2 Utilization rate and conversion rate of H2O2
2.2.5 反应温度的影响
实验条件:2 mmol 1-己烯,2 mmol 30% H2O2,0.05 g Co-WOx-0.1,8 h,n(H2O2:1-己烯)=1。如图8(c)所示,环氧化反应最适宜的温度是60 ℃。当温度低于60 ℃时,分子的扩散速率减慢,反应速率降低。较低温度有利于1-己烯在催化剂表面的吸附,然而不利于活性中间体中活性氧向1-己烯双键转移[14],部分吸附态的1-己烯被直接氧化成正戊醛;另一方面,生成的1,2-环氧己烷因在低温下不易脱附,所以在表面B酸的作用下开环生成了1,2-己二醇,并进一步发生过度氧化生成2-羰基-1-己醇。同时,Co-WOx-0.1的活性位点吸附1-己烯与其活化H2O2存在竞争关系,不易脱附的1-己烯占据部分活性位点,活化H2O2的位点会减少,1,2-环氧己烷的产量降低。然而,温度过高不仅会加速吸热的水解反应发生,促使1,2-环氧己烷生成1,2-己二醇,从而导致1,2-环氧己烷的选择性下降,温度升高还会加速H2O2直接分解,降低H2O2的利用率[46]。
综合以上实验,在本工作中Co-WOx-0.1催化剂的最佳反应条件是2 mmol 1-己烯,2 mmol 30%H2O2,催化剂用量0.05 g,60 ℃,8 h,n(H2O2∶1-己烯) = 1,在此条件下,1-己烯的转化率为39.8%,1,2-环氧己烷的选择性为55.7%。
2.3 反应机理的分析
从前面实验数据可知,Co-WOx-0.1催化剂上1,2-环氧己烷的选择性是明显升高的,但是其转化率比纯WOx转化率略低5.3%左右。如前所述,WOx表面B酸含量是影响1,2-环氧己烷选择性的主要原因。在WOx体系中,8 h反应后1,2-环氧己烷的选择性仅为26.9%,而1,2-己二醇和2-羰基-1-己醇两者之和达到45.2%,其均与1,2-环氧己烷的开环水解作用有关。根据反应平衡可知,产物1,2-环氧己烷的不断水解有利于化学平衡正向进行,从而增加了1-己烯的转化。然而,在Co-WOx-0.1体系中,1,2-环氧己烷的水解作用被大幅度抑制,使得反应中底物生成1,2-环氧己烷的反应也受到抑制,从而降低了1-己烯的转化率。
根据文献报道,WOx体系环氧化的活性位点是其表面的氧空位[24]。一方面,1-己烯双键具有π电子,氧空位本身是电子受体,因此,氧空位对1-己烯有吸附作用;另一方面,H2O2会在氧空位处被活化从而和W5+形成W-O-OH活性结构[14,47],其传递一个活性氧给1-己烯双键从而实现环氧化过程。本工作中,Co2+增加了WOx表面氧空位,最大可能原因是Co2+对其晶格原子产生了占位作用。因为Co-O键长(2.13 Å)均大于W-O在a、c平面和b方向上的最大键长2.03和1.89 Å,且O-Co-O的键角也不同于O-W-O的键角,因此,Co2+的掺杂很容易引起W-O配位的不饱和,从而产生氧空位。通过文献调研发现,无论是钛基催化剂催化烯烃环氧化体系[48-50],还是以过渡金属氧化物(W、Nb、Ta等)为催化剂的环氧化反应[47,51],其反应活性中间体均为M-O-OH。结合Co-WOx-0.1具有氧空位可以活化H2O2这一特点,推测了以W-O-OH为中间体的反应机理。
WOx催化剂催化环氧化过程如图8(d)所示。首先,H2O2向Co-WOx-0.1中的氧空位进行亲核加成[24],形成W(O2)三元环结构(图8(d)(1));该三元环结构张力较大极不稳定,在H2O2和催化剂表面W-OH电离形成的H+作用下,迅速水解形成W-OOH结构(图8(d)(2));第三步,W-O-OH向1-己烯的C=C键提供活性氧形成吸附态的1,2-环氧己烷,并失去一分子H2O(图8(d)(3));最后1,2-环氧己烷于催化剂表面脱附,完成催化反应循环(图8d(4))。该催化循环中还有其他副反应发生:一是未被活化的H2O2分解生成O2和H2O[52];二是α-烯烃烯丙基位置的氢比较活泼易被氧化[53,54],生成可以稳定存在的2-羟基-1-己烯和2-羰基-1-己烯;三是1,2-环氧己烷在表面B酸的作用下,开环水解生成1,2-己二醇,因为仲碳位置的羟基比较活泼,可被进一步氧化生成具有p-π共轭效应的2-羰基-1-己醇;四是少部分烯烃双键直接被氧化成正戊醛,正戊醛可以再进一步被氧化生成正戊酸。
3 结 论
本研究以动态溶剂热法制备了Co掺杂的Co-WOx-x催化剂(x= 0.05、0.1和0.2),Co2+的引入减少了WOx表面B酸含量,同时增加了表面氧空位的含量。在1-己烯的环氧化反应中,和纯WOx相比,Co-WOx-0.1催化剂在1-己烯转化率降低约5.3%情况下将1,2-环氧己烷的选择性提升了28.8%,明显提升了WOx环氧化性能。本研究系统考察了反应参数对环氧化过程的影响,并对Co-WOx催化剂相关环氧化机理做了深入讨论。
