HPLC法同时测定木香顺气丸中6种成分
2022-12-05李静华王丽琼
李静华, 王丽琼
(1.乐山职业技术学院,四川 乐山 614000;2.乐山市食品药品检验检测中心,四川 乐山 614000;3.四川省药品监督管理局中药质量研究重点实验室,四川 乐山 614000)
木香顺气丸功效行气化湿、健脾和胃,方中厚朴、枳壳、陈皮、青皮为臣药,甘草为使药[1],其中甘草指标成分为甘草苷,健脾益气,补中除湿[2-3];陈皮、青皮、枳实指标成分为柚皮苷、橙皮苷、新橙皮苷[4-6],行气燥湿,散结消积;厚朴指标成分为和厚朴酚、厚朴酚[7-8],行气燥湿,散结消积。该制剂收载于2020年版《中国药典》一部,但只测定了和厚朴酚、厚朴酚含量[9],而中药质量和疗效提倡整体性,故多组分同时测定可更全面地体现其质量[10-12]。
前期孙全明[13]、曹瑞竹[14]等采用HPLC法测定木香顺气丸中木香烃内酯、去氢木香烃内酯、厚朴酚、和厚朴酚、甘草酸含量,王玲娇等[15]采用光谱指纹法评价木香顺气丸质量,但尚无同时测定甘草苷、柚皮苷、橙皮苷、新橙皮苷、和厚朴酚、厚朴酚含量的报道。因此,本实验建立HPLC法同时测定该制剂中上述6种成分的含量,以期为整体全面地控制其质量提供可靠科学的依据。
1 材料
Agilent 1260高效液相色谱仪,配置二极管阵列检测器(美国Agilent公司);XS205电子天平(瑞士梅特勒-托利多公司);KQ5200DB超声波清洗器(昆山市超声仪器有限公司);ULUP-II-10T优普系列超纯水器(成都超纯科技有限公司)。
厚朴酚(批号110729-201513,纯度98.8%)、和厚朴酚(批号110730-201614,纯度99.3%)、新橙皮苷(批号111857-201804,纯度99.4%)、橙皮苷(批号110721-201818,纯度96.2%)、柚皮苷(批号110722-201613,纯度94.3%)、甘草苷(批号111610-201607,纯度93.1%)对照品均购自中国食品药品检定研究院。木香顺气丸(甘肃佛仁制药科技有限公司,批号200401;山西万辉制药有限公司,批号191101;甘肃河西制药有限责任公司,批号200901),规格均为每袋6 g。乙腈、甲醇为色谱纯(北京百灵威科技有限公司);N-N二甲基甲酰胺为分析纯(成都市科隆化学品有限公司);磷酸为分析纯[重庆川东化工(集团)有限公司];水为纯化水。
2 方法与结果
2.1 溶液制备
2.1.1 对照品溶液 精密称取对照品厚朴酚24.78 mg、和厚朴酚14.19 mg、新橙皮苷17.50 mg、橙皮苷120.62 mg、柚皮苷27.61 mg、甘草苷11.04 mg,分别置于6个25 mL量瓶中,N-N二甲基甲酰胺溶解并稀释至刻度,摇匀,分别精密量取5、5、10、10、10、2 mL,置于同一50 mL量瓶中,N-N二甲基甲酰胺稀释至刻度,摇匀,作为贮备液,精密量取5 mL,置于10 mL量瓶中,N-N二甲基甲酰胺稀释至刻度,摇匀,即得。
2.1.2 供试品溶液 将本品粉碎,取约1 g,置于具塞锥形瓶中,精密加入N-N二甲基甲酰胺50 mL,称定质量,超声处理1 h,放冷,N-N二甲基甲酰胺补足减失的质量,摇匀,0.45 μm微孔滤膜过滤,取续滤液,即得。
2.1.3 阴性样品溶液 按处方制得不含陈皮、青皮、枳壳、甘草、厚朴的阴性样品,按“2.1.2”项下方法制备,即得。
2.2 色谱条件 SVEA C18Opal色谱柱(250 mm×4.6 mm,5 μm);流动相0.1%磷酸(A)-乙腈(B)-甲醇(C),梯度洗脱,程序见表1;体积流量1.0 mL/min;柱温30 ℃;检测波长276 nm;进样量10 μL。
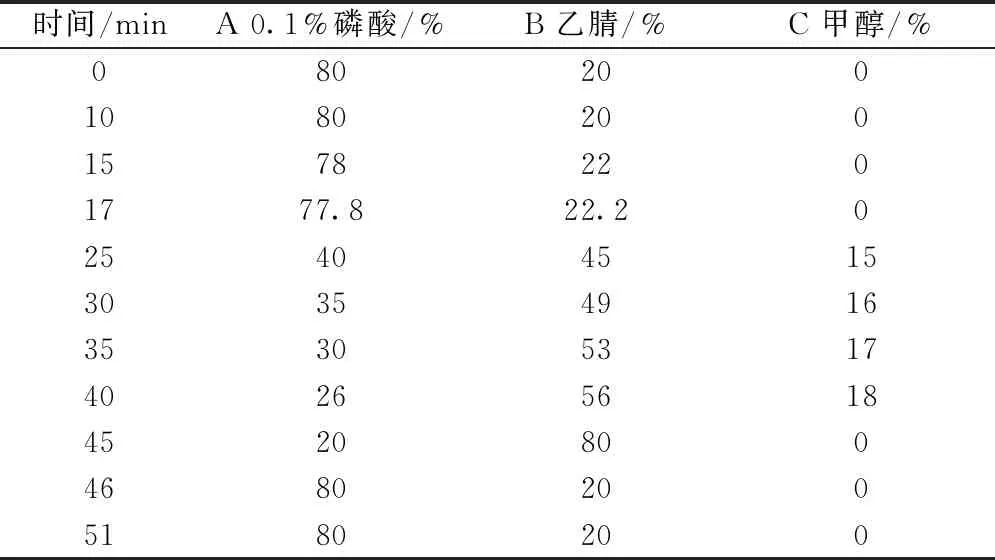
表1 梯度洗脱程序
2.3 专属性考察 取对照品、供试品、阴性样品溶液适量,在”2.2”项色谱条件下进样测定,结果见图1。由此可知,供试品溶液色谱图中各成分保留时间与对照品溶液色谱图中一致,并且分离度符合要求,阴性无干扰,表明该方法专属性良好。
2.4 线性关系考察 精密吸取“2.1.1”项下贮备液8、6、4、2、1 mL,分别置于5个10 mL量瓶中,N-N二甲基甲酰胺稀释至刻度,摇匀,在“2.2”项色谱条件下进样测定。以对照品质量浓度为横坐标(X),峰面积为纵坐标(Y)进行回归,再以S/N=10计算定量限,结果见表2,可知各成分在各自范围内线性关系良好。
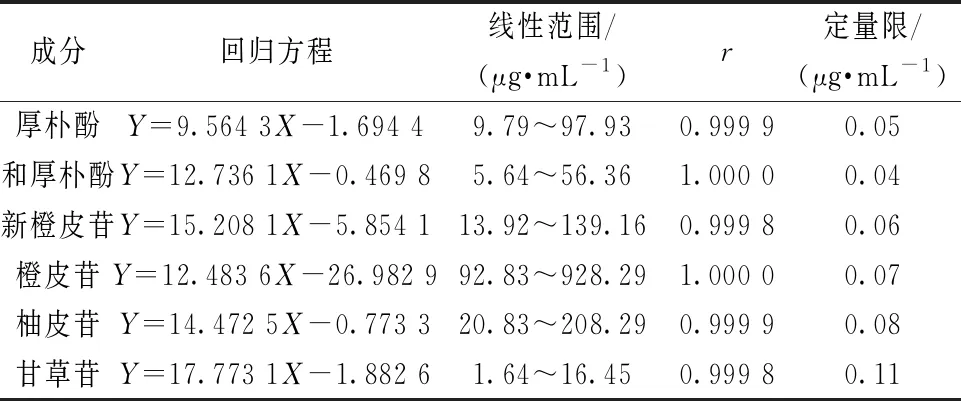
表2 各成分线性关系
2.5 精密度试验 取“2.1.1”项下对照品溶液适量,在“2.2”项色谱条件下进样测定6次,测得厚朴酚、和厚朴酚、新橙皮苷、橙皮苷、柚皮苷、甘草苷峰面积RSD分别为0.4%,0.3%,0.7%,0.8%,1.1%、0.9%,表明仪器精密度良好。
2.6 重复性试验 取同一批本品(批号200401),按“2.1.2”项下方法平行制备6份供试品溶液,在“2.2”项色谱条件下进样测定,测得厚朴酚、和厚朴酚、新橙皮苷、橙皮苷、柚皮苷、甘草苷含量RSD分别为1.2%,1.3%,1.6%,1.6%,1.9%、1.9%,表明该方法重复性良好。
2.7 稳定性试验 取同一份供试品溶液(批号200401)适量,室温下于0、2、4、8、12、24 h在“2.2”项色谱条件下进样测定,测得厚朴酚、和厚朴酚、新橙皮苷、橙皮苷、柚皮苷、甘草苷峰面积RSD分别为0.9%、0.9%、1.2%、1.3%、1.4%、1.1%,表明溶液在24 h内稳定性良好。
2.8 加样回收率试验 取各成分含量已知的本品(批号200401,厚朴酚、和厚朴酚、新橙皮苷、橙皮苷、柚皮苷、甘草苷含量分别为2.089 2、0.752 7、1.443 7、20.935 5、1.935 4、0.148 5 mg/g)6份,每份约0.5 g,精密称定,置于具塞锥形瓶中,精密加入 “2.1.1”项下0.2 mL甘草苷贮备液、1 mL柚皮苷贮备液、2 mL橙皮苷贮备液、1 mL新橙皮苷贮备液、0.7 mL和厚朴酚贮备液、1 mL厚朴酚贮备液,按“2.1.2”项下方法制备供试品溶液各6份,在“2.2”项色谱条件下各进样10 μL测定,计算回收率。结果,厚朴酚、和厚朴酚、新橙皮苷、橙皮苷、柚皮苷、甘草苷平均加样回收率分别为101.0% (RSD=2.0%)、100.7% (RSD=1.5%)、100.1% (RSD =1.4%)、99.8% (RSD =1.7%)、100.6% (RSD =1.9%)、99.1%(RSD=1.9%)。
2.9 样品含量测定 取3批样品,按“2.1.2”项下方法制备供试品溶液,每批平行3份,在“2.2”项色谱条件下进样测定,外标法计算含量,结果见表3。
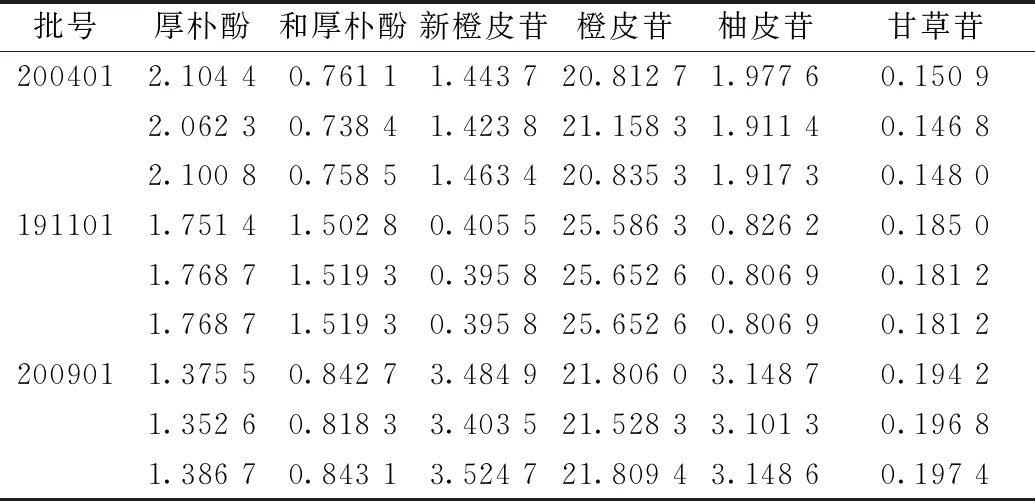
表3 各成分含量测定结果(mg/g,n=3)
3 讨论
3.1 检测波长选择 本实验发现,甘草苷在276 nm波长处有吸收峰,而且吸收强度较大,同时厚朴酚、和厚朴酚、新橙皮苷、橙皮苷、柚皮苷在该处的吸收强度也较好。最终确定,检测波长为276 nm。
3.2 流动相选择 本实验比较了乙腈、甲醇对各成分分离效果的影响,发现采用乙腈时色谱峰保留时间提前,峰形尖锐,并且加入一定比例甲醇后保留时间适中,分离度良好。由于各成分理化性质不一致,故前10 min进行大比例水相等度洗脱,10~17 min进行梯度洗脱,可将柚皮苷、橙皮苷与相邻杂质峰的分离度提高到3.0左右,从而排除干扰峰的影响。
3.3 提取溶剂选择 本实验比较了50%甲醇、2%醋酸甲醇、甲醇、N-N二甲基甲酰胺、甲醇-(N-N二甲基甲酰胺)混合溶液(比例分别为45∶5、40∶10、30∶20、20∶30、10∶40、5∶45)对各成分提取能力的影响,发现甘草苷、柚皮苷、橙皮苷、新橙皮苷提取率受溶剂种类的影响较大,尤其是含量较高的橙皮苷;以N-N二甲基甲酰胺提取时,各成分溶解性较好,提取完全。最终确定,提取溶剂为N-N二甲基甲酰胺。
4 结论
本实验采用HPLC法同时测定木香顺气丸中厚朴酚、和厚朴酚、新橙皮苷、橙皮苷、柚皮苷、甘草苷的含量,该方法操作简单,方法学考察结果良好,可为整体全面地控制该制剂质量提供可靠科学的依据。
