聚吡咯/环氧树脂复合材料的制备及其性能研究
2022-12-01习绍华AbdulWariz陈卓然朱建锋
郭 江, 习绍华, Abdul Wariz, 李 帅, 陈卓然,李 旭, 王 芬, 朱建锋
(陕西科技大学 材料科学与工程学院 陕西省无机材料绿色制备与功能化重点实验室, 陕西 西安 710021)
0 引言
随着科技的不断发展,基于电磁波信息传输的各类设备和系统的使用日渐频繁,然而其产生的电磁波干扰、辐射已经严重威胁到军事安全和人类健康[1].为了消除电磁波负面危害、有效控制电磁污染,尽量减少辐射污染对仪器设备的干扰,并使人体处于安全辐射范围,研发高效优异的电磁波吸收材料势在必行[2,3].电磁波吸收性能主要取决于阻抗匹配和衰减特性,但是单组分电磁波吸收材料难以满足“薄、轻、强、宽”的发展要求.因此,优化复合材料的电磁波吸收性能是目前的研究热点[4,5].
近年来,导电聚合物及其纳米复合材料因其用途广泛、重量轻、耐腐蚀性好、导电性可调等优点,引起了科学界和工业界的关注[6-8].其中聚吡咯(PPy)作为导电聚合物之一,由于具有高导电性、环境稳定性、易于合成以及独特的氧化还原特性等已被广泛应用于吸波材料、电磁屏蔽材料等领域[9,10].Su等[11]通过简单工艺制备了新型石墨纳米片/核壳Fe3O4@PPy微波吸收材料,该复合材料(厚度1.5 mm)在9.92 GHz下达到最大反射-49.3 dB. Ge等[12]通过原位聚合过程成功制备ZnFe2O4@PPy纳米复合材料,其表现出优异的电磁吸波性能(厚度2.5 mm、频率30.24 GHz下达到-42.31 dB)和吸收带宽(9.66~37.86 GHz).虽然PPy有利于增强导电聚合物型复合材料对电磁波的电损耗[13],但其较差的力学性能限制了实际工程应用中的价值.而当前许多吸波类工程应用材料包括军用飞机、吸波建筑材料、实验室屏蔽设备等均需具备优异的力学性能以抵抗外界环境的损害。因此,设计同时具有优异电磁波吸收性能和机械性能的复合材料至关重要.
环氧树脂是应用最广泛的工程热固性材料之一,具有力学性能高、粘结性能优异、固化收缩率小、工艺性能好等优点[14,15],其可以一定程度上改善导电聚合物的机械性能,因此导电聚合物/环氧树脂复合材料在工程应用的电磁吸波方面具有很大发展潜力.如Guo等[16]研究了不同含量的聚苯胺填料对聚苯胺/环氧树脂复合材料吸波性能的影响,结果表明10.0 wt% 聚苯胺/环氧树脂在17.7 GHz时的反射损耗达到-36.8 dB.另外,本课题组在先前的工作中[17]证明了PPy可以通过C-N共价键与环氧树脂发生反应,所形成的共价键使聚吡咯均匀分散在环氧基体中,从而形成导电网络.然而环氧树脂是天然的绝缘体,不能吸收或反射电磁波,电磁波可以很容易地进入或离开壳体。因此探究何种复合方式能使得最终获得的PPy/环氧树脂复合材料既具有PPy优良的电性能与电磁屏蔽性能又具有环氧树脂优异的工程实用性是本实验的关键.
针对上述内容,本实验采用氧化模板法制备纤维PPy,利用浸润浇铸的方法将纤维PPy与环氧树脂进行复合,通过改变纤维PPy的掺杂量,系统研究了PPy填料对材料力学性能和吸波性能的影响.本实验可为处理电磁波污染的环氧树脂基复合材料的制备提供技术指导.
1 实验部分
1.1 实验原料
吡咯、十六烷三甲基溴化铵(CTAB)、过硫酸铵(APS)、无水乙醇,购自天津科密欧化学试剂有限公司;环氧树脂862,购自广州市镐韵化工有限公司;二乙基甲苯二胺,购自济宁百川化工有限公司;盐酸,购自天津科天力化学试剂有限公司.以上试剂均为分析纯试剂.去离子水采用实验室超纯水一体机自制.
1.2 纤维聚吡咯(PPy)的制备
纤维聚吡咯的制备过程如图1所示.称取1.13 g过硫酸铵(APS)溶于200 mL去离子水中,标记为溶液A.称取0.72 g十六烷基三甲基溴化铵(CTAB)溶于200 mL的1 mol/L HCl溶液中,同时冰浴(0 ℃~5 ℃)搅拌20 min,搅拌结束后取344 mL(即0.33 g),并将吡咯单体加入其中,搅拌30 min,作为溶液B.在0 ℃~5 ℃冰浴条件下将溶液A缓慢滴入溶液B中反应24 h.聚合反应完成后,进行真空抽滤或离心收集其沉淀物,用水和无水乙醇洗涤,除去过量的酸、多余的低聚物和额外的有机溶剂.将得到的沉淀物在80 ℃下干燥12 h,即得到纤维聚吡咯.
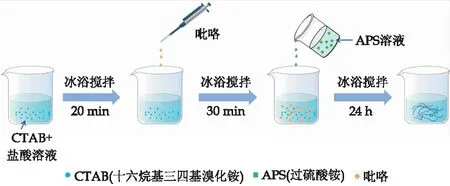
图1 纤维聚吡咯聚吡咯制备流程图
1.3 PPy/环氧树脂复合材料的制备
采用浸润浇铸法制备纤维PPy/环氧树脂复合材料.在开始实验前需对模具进行处理:使用无水乙醇擦拭模具,去除表面杂物,涂抹脱模剂后放入干燥箱中80 ℃干燥3 h,随后在真空干燥箱中120 ℃真空干燥1 h.其制备过程如图2所示.
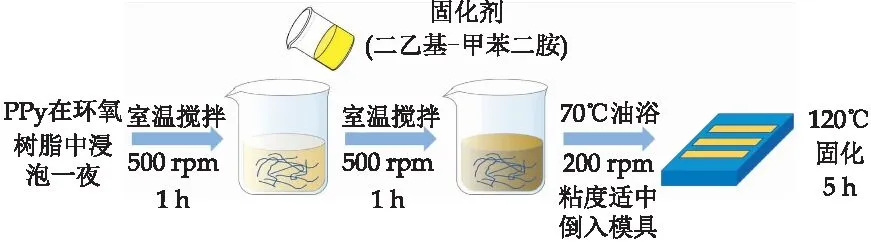
图2 PPy/环氧树脂复合材料的制备
首先,将纤维PPy浸泡在环氧树脂中12 h,使PPy完全浸润在环氧树脂中,将混合物在室温下机械搅拌(500 rpm)1 h.然后以环氧树脂单体/固化剂重量比为100/26.5 (环氧树脂和固化剂的总质量25.3 g)的比例加入固化剂,机械搅拌1 h,转速500 rpm.随后升温至70 ℃,降低转速至200 rpm,观察粘度,当粘度适中时倒入模具中,在120 ℃烘箱中固化5 h,自然冷却至室温,即得到所制备的PPy/环氧树脂复合材料.
采用上述方法,本实验制备了PPy含量为1 wt%、3 wt%、6 wt%的三种PPy/环氧树脂纳米复合材料,并用相同方法制备了纯环氧树脂用来作性能对比.
1.4 测试与表征
试样晶体结构分析采用日本Rigaku公司D/Max-2200pc型X射线衍射仪(XRD),其中管压:40 kV,管流:40 mA,阳极靶材为Cu靶(λ=0.154 18 nm),扫描范围:15°~70°,扫描速度:2 °/min. 试样表面微观结构通过日本日立公司S-4800型场发射扫描电子显微镜进行观察,电子加速电压控制在3~5 kV.试样表面化学和结构分析采用傅立叶红外光谱分析仪(Vertex 70)和拉曼光谱分析仪.PPy/环氧树脂纳米复合材料的拉伸测试采用AI-7000GD单向拉伸试验机,其中实验所用样品为哑铃型,夹长:25 mm,标距:15 mm,拉伸速度:1 mm/min.
2 结果与讨论
2.1 纤维聚吡咯(PPy)的微观形貌与晶体结构分析
图3为纤维PPy的SEM微观形貌图.由图3可以看出,通过氧化模板法制备出的聚吡咯为纤维状,纤维表面光滑,无明显的团聚现象.纤维PPy纳米结构的直径较为均匀,具有较高的长径比,说明纤维PPy具有较大的比表面积,当其作为填料加入环氧树脂基体时,与基体的结合力会更强,也更容易在基体中形成网络状结构.
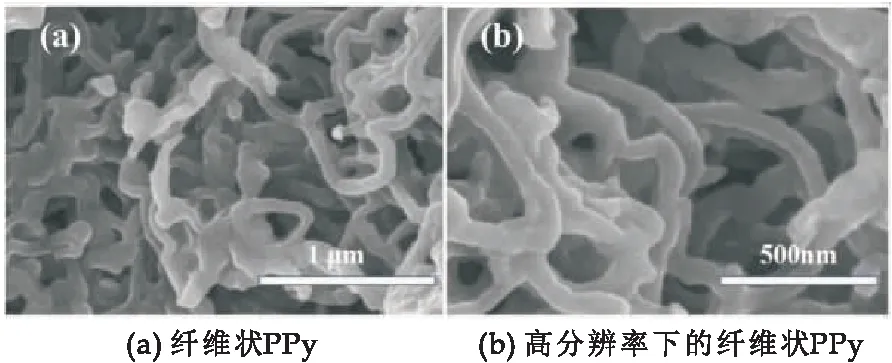
图3 PPy的SEM图
此外,实验中发现,在过硫酸铵滴入盐酸、CTAB和吡咯单体混合溶液的过程中,少量滴入时会出现一种白色的沉淀物,然后随着过硫酸铵滴入量的增加沉淀物变为黑色.这种白色絮状沉淀物的出现与文献报导的实验现象类似[18].分析其原因得出:随着吡咯单体的加入,CTAB/HCl/吡咯单体混合溶液的粘度增大,粘度较高时会促进PPy纳米线的形成,因此CTAB/HCl/吡咯体系可以为吡咯单体形成纤维聚吡咯提供所需的模板.
图4为盐酸掺杂的PPy的X射线衍射图谱.对于纯PPy,衍射峰2θ=22.00°~30.00°之间出现明显的衍射宽峰,这是因为PPy的无定型所致.图5是纳米纤维聚吡咯的傅里叶红外光谱图,图中所示在波长为1 548 cm-1和1 481 cm-1分别归属于聚吡咯环中C=C的伸缩振动峰和吡咯环的C-C伸缩振动吸收峰[19],1 298 cm-1和1 172 cm-1处的峰分别是由C-N的伸缩振动和吡咯环上C-N的伸缩振动引起的[20,21],位于962 cm-1~1 045 cm-1处的吸收峰表示了C-H和N-H键在吡咯环内的变形振动[22].通过FTIR分析进一步证明聚吡咯被成功合成.

图4 纤维聚吡咯的XRD图谱
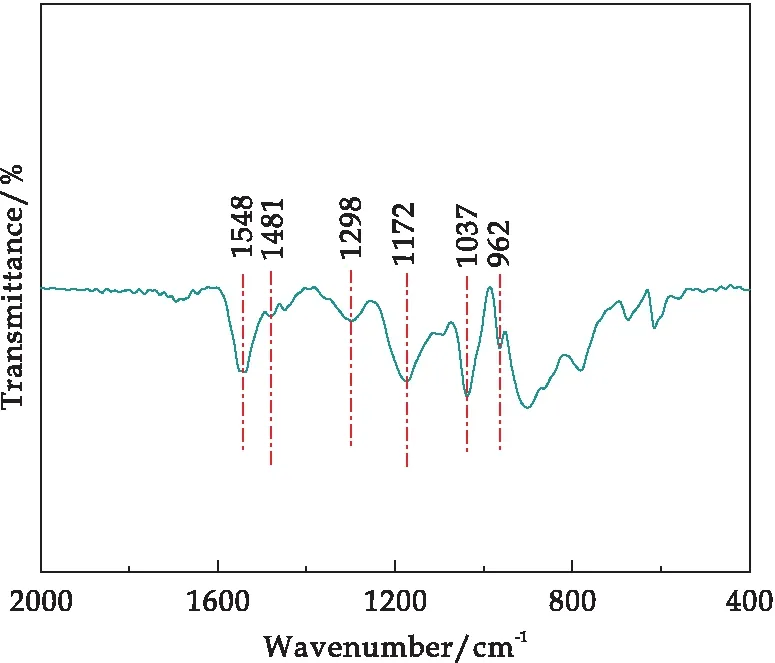
图5 纤维聚吡咯的红外光谱图
2.2 PPy/环氧树脂复合材料的性能测试
图6为加入不同PPy填料的环氧树脂纳米复合材料的应力-应变曲线.3 wt% PPy/环氧树脂纳米复合材料的拉伸强度最高,达到80.1 MPa,高出纯环氧树脂的65.0 MPa.随着PPy填料含量增加至6 wt%时,PPy/环氧树脂纳米复合材料的拉伸强度显著降低至67.9 MPa,这是由于PPy含量过高,引起了PPy纳米纤维的团聚,使之在环氧树脂中的分散性变差,从而降低了拉伸强度.
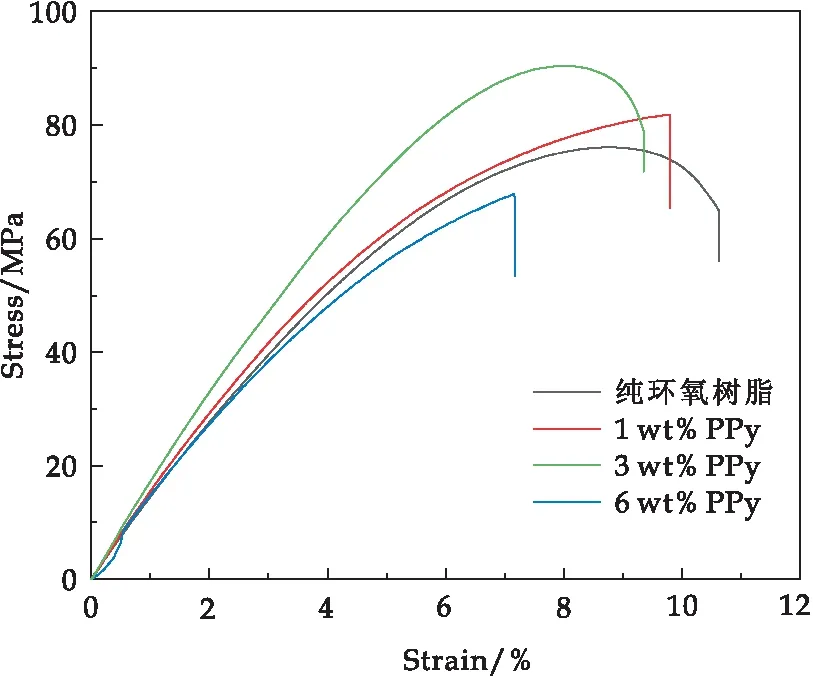
图6 PPy/环氧树脂复合材料的应力-应变曲线
根据PPy/环氧树脂复合材料的拉伸数据可以计算出其杨氏模量(如图7所示),杨氏模量先是随着PPy含量的增加而递增,在PPy含量为3 wt%时达到最大值,之后开始呈现下降趋势.杨氏模量越大,材料抵抗形变的能力越强,刚性越好.由此结果可知,在PPy填料含量为3 wt%时,PPy/环氧树脂复合材料杨氏模量最大,使其在外加力的作用下不容易发生形变,刚性最好;但PPy含量不断增大会使PPy产生团聚现象,并且和环氧树脂基体的结合性变差,抵抗外力形变的能力下降,杨氏模量下降,刚性变差.
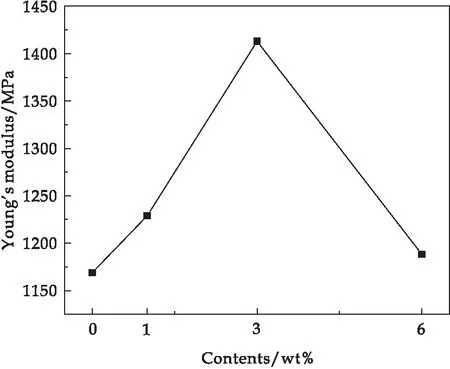
图7 PPy/环氧树脂复合材料的杨氏模量
纯环氧树脂以及PPy填料含量不同的PPy/环氧树脂复合材料拉伸测试后其拉伸断面的微观形貌如图8所示。在图8(a)中,纯环氧树脂的断面比较光滑,这是由于脆性断裂时裂纹的快速扩展造成的.与纯环氧树脂相比,不同含量PPy填料的复合材料断裂表面更加粗糙,这是由于基体的剪切应力与PPy纳米纤维相互作用而形成的.从图8(a)、(b)、(c)、(d)可以看出,随着PPy填料含量的不断增大,PPy/环氧树脂复合材料显示出越来越粗糙的断面形貌,断面上的PPy也越来越明显.且在PPy填料含量为3 wt%和6 wt%的PPy/环氧树脂复合材料的断面均观察到团聚现象.团聚的原因可能是:聚吡咯纳米结构的尺寸较小、比表面积较大以及其表面原子具有较大的活性,所以在PPy填料含量较高时,容易产生团聚现象.

图8 PPy/环氧树脂复合材料的拉伸断面SEM图
通过测试PPy/环氧树脂复合材料的体积电阻率,结果显示随着PPy含量的上升而降低,出现这种现象的原因是由于制备的纤维PPy具有较高的长径比和优良的导电性能,随着PPy含量的增加,PPy纳米结构互相连通形成导电网络,使复合材料的导电性能提高,体电阻率降低.
材料的吸波性能取决于复介电常数和复磁导率[23].复介电常数的实部(ε′)和虚部(ε″)分别代表介质电能的储存和损耗.PPy/环氧树脂复合材料在2~18 GHz范围内的复介电常数如图9所示.
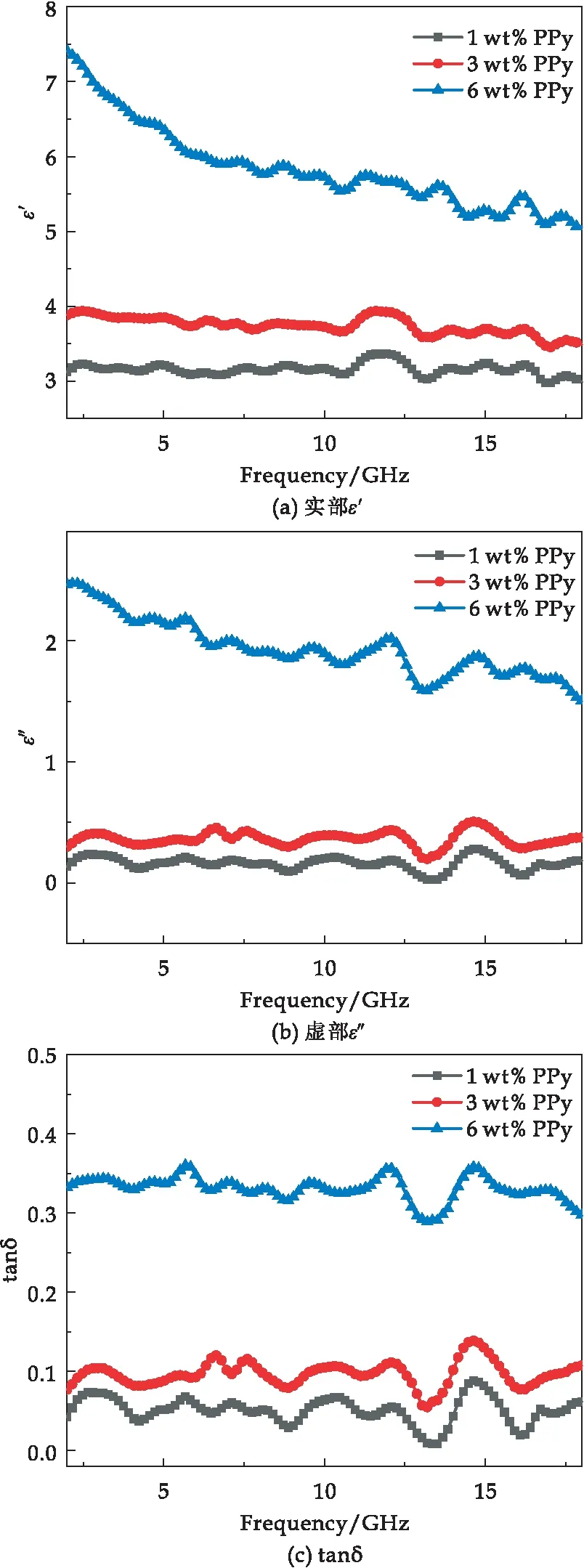
图9 PPy/环氧树脂复合材料的复介电常数
PPy/环氧树脂复合材料的ε′均为正值,这是由于电荷载流子的移动受到环氧树脂的阻碍,导致电荷载流子在PPy和环氧树脂之间的界面处积聚,使得PPy和环氧树脂之间的界面处形成界面极化[24].在图9(a)、(b)中,ε′在相同频率下随着PPy含量的增加而增加,这是由于PPy填料在复合材料中的占比增加,与环氧树脂的接触面积增加,从而导致更强的界面极化效应.同样,复介电常数的ε″有相同的变化趋势.
反射损耗(RL)可以按公式(1)计算[25]:
(1)
Zin为有效输入阻抗,计算公式如式(2)[26]:
(2)
式(2)中:εr和μr分别为吸收体的复介电常数和渗透率,d为吸收层的厚度,c为光速,f为电磁波频率,μr=μ′-jμ″和εr=ε′-jε″分别是复合材料的相对磁导率和介电常数.
一般来说,当RL值低于-10 dB时,能有效吸收90 %以上的入射电磁波,定义RL<-10 dB的频带为材料的有效吸收带宽.
在不同厚度下,不同PPy填料含量的PPy/环氧树脂复合材料的RL如图10所示.
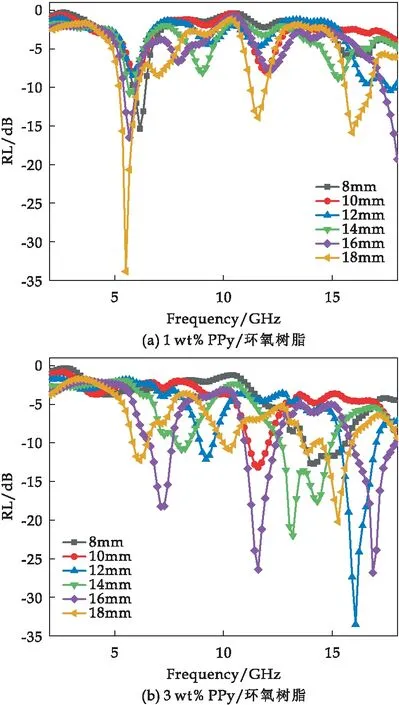
图10 PPy/环氧树脂复合材料的吸波性能
由图10(b)可以看出,3 wt% PPy添加量下,匹配厚度为12 mm时,PPy/环氧树脂纳米复合材料在16.1 GHz下的最小反射损耗RL达到-33.50 dB,有效吸收带宽为1.56 GHz(15.44~17 GHz).而6 wt% PPy/环氧树脂复合材料吸波性能最差,这是由于PPy填料含量较大,过高的介电常数使得复合材料的反射增强,阻抗匹配变差,影响其吸波性能[27].
3 结论
本文以环氧树脂为基体,纤维PPy为填料,成功制备出了PPy/环氧树脂复合材料.3 wt%PPy/环氧树脂复合材料的拉伸强度达到80.1 MPa.此外,在PPy与环氧树脂之间形成界面层,这提高了PPy/环氧树脂复合材料的杨氏模量.同时,3 wt%PPy/环氧树脂纳米复合材料也表现出良好的电磁吸收性能,在16.1 GHz下的最小反射损耗RL达到-33.50 dB,这是由于PPy填料增加了材料内部的多次反射.因此,PPy/环氧树脂纳米复合材料具有良好的机械性能和电磁波吸收性能,有望成为一种先进的多功能材料.
