基于组蛋白修饰数据预测基因差异性表达的深度融合模型
2022-11-30李昕贾韬
李昕,贾韬
基于组蛋白修饰数据预测基因差异性表达的深度融合模型
李昕,贾韬*
(西南大学 计算机与信息科学学院,重庆 400715)(∗通信作者电子邮箱tjia@swu.edu.cn)
针对使用大规模组蛋白修饰(HM)数据预测基因差异性表达(DGE)时未合理利用细胞型特异性(CS)和细胞型间异同两类信息,且输入规模大、计算量高等问题,提出一种深度学习方法dcsDiff。首先,使用多个自编码器(AE)和双向长短时记忆(Bi‑LSTM)网络降维,并建模HM信号得到嵌入表示;然后,利用多个卷积神经网络(CNN)分别挖掘每类CS的HM组合效应以及两细胞型间每种HM的异同信息和所有HM的联合影响;最后,融合两类信息预测两细胞型间的 DGE。在对REMC数据库中10对细胞型的实验中,与DeepDiff相比,dcsDiff的预测DGE的皮尔逊相关系数(PCC)最高提升了7.2%、平均提升了3.9%,准确检测出差异表达基因的数量最多增加了36、平均增加了17.6,运行时间节省了78.7%;进一步的成分分析实验证明了合理整合上述两类信息的有效性;并通过实验确定了算法的参数。实验结果表明dcsDiff能有效提高DGE预测的效率。
组蛋白修饰;基因差异性表达;细胞型特异性;自编码器;双向长短时记忆网络;信息融合;表观遗传学
0 引言
组蛋白修饰(Histone Modification, HM)是最重要的表观遗传分子机制之一,指组蛋白在相关酶作用下的修饰过程,如甲基化、乙酰化、磷酸化、腺苷化、泛素化等[1]。这些修饰涉及不同的脱氧核糖核酸(DeoxyriboNucleic Acid, DNA)区域(例如启动子区域和增强子区域)[2-3],通过改变或限制DNA相关区域的可接近性,HM可以激活或抑制特定功能基因的表达,从而影响生命过程[4]。HM谱的变化可能导致基因的差异性表达(Differential Gene Expression, DGE)现象[5-6],即特异性基因在不同细胞或同一细胞的不同发育阶段出现了表达差异,从而产生特异性蛋白,导致细胞形态、结构和功能的差异[7-9]。DGE与各种生物表型和许多疾病的遗传过程密切相关。通过分析不同细胞型间的HM模式,探索HM模式对基因表达(Gene Expression, GE)的影响,预测DGE并检测差异表达的基因,可以为复杂疾病的研究和表观遗传药物的开发提供新的见解。
人们在GE领域做出了许多努力以理解其分子机制[10-20]。为预测GE,研究人员尝试使用各种类型的生物学数据,包括转录因子结合位点(Transcription Factor Binding Site, TFBS)数据[21]、DNA甲基化数据[22]、DNA可及性数据[23]、组蛋白修饰数据[24],还有一些融合多个数据源[25-30]。然而,文献[26]中证明了转录因子结合位点数据和HM数据在预测GE方面具有严格的统计学意义上的冗余性。再者,HM可以调节染色质的开放程度来影响DNA的可及性。当DNA可及性较高时,后续的转录因子可与DNA结合,从而影响GE。因此,仅利用多种多样的HM数据就能准确预测GE。鉴于此,目前已有多种计算方法通过挖掘大规模HM信号[31-33]来预测GE,可以降低分析大规模HM信号的传统实验的高昂成本。
最早的方法是将整个选定的基因区域内HM信号的平均值作为线性回归和规则学习的输入特征[24-34]。然而,不同基因各自的HM信号在DNA上具有不同的分布模式,这种方法默认所有基因的所有HM都是相同的分布模式,无法捕捉到分布差异信息,影响了对目标GE的预测精度。接着binning策略应运而生,即把每个基因以转录起始位点(Transcription Start Site, TSS)为中心的DNA区域划分成更小的箱子(记作bin),然后提取每个bin的HM信号作为机器学习模型(如支持向量机、支持向量回归(Support Vector Regression, SVR)、随机森林)的输入特征,以预测GE[26,35-36]。早期基于binning策略的方法可以进一步分为两种类型:特异性bin和最佳bin方法。特异性bin方法通常分开为每个bin建模,而最佳bin方法仅选择与GE相关性最大的一个bin作为输入。这些早期的binning方法可以识别不同HM信号分布对GE的差异影响,但忽略了输入bin之间的连接关系,导致重要的表观遗传信息丢失。基于深度学习的方法可以克服早期binning方法的不足。深度学习方法可以对输入HM信号进行非线性编码,捕获输入样本的局部和全局特征,并能自动挖掘输入bin之间的连接性与HM间的组合效应[37-39]。尽管深度学习方法改进了利用HM信号进行GE预测的方法,但它们仍然存在一些问题。首先,它们或没有考虑HM的模块化特性(即每种HM可以被视为一个小的遗传模块),没有分别为每类HM建模,而是直接简单将一个基因所有类型的HM原始信息一并融合处理;或在综合捕获各HM组合效应时的粒度不够精细,只是在宏观上考量各HM组合起来的影响,没有在DNA区域每一个局部位置考察该处各HM的局部组合效应;再者,不同组织类型或不同细胞发育阶段的GE是不同的。当需要分析基因在两种细胞型之间发生差异性表达的原因时,细胞型特异性(Cell type‑Specificity, CS)基因表达预测方法只能分别预测各单一细胞型下的GE值,然后基于GE值计算不同细胞型之间的DGE值,这种预测DGE的方法是不连贯且孤立的,更不能捕获每个基因在不同细胞型中HM的差异信息。采用一致性方法直接预测两种细胞型间的DGE值比细胞型特异性基因表达(CS GE)预测更准确可靠,但大多数方法仍侧重于单一细胞型的CS GE预测,DGE预测方法仍比较缺乏。
考虑到这些问题,本文提出了dcsDiff算法,基于深度学习分别对不同细胞型间HM的异同信息和单一细胞型下的HM特异性信息进行针对性建模,并将其融合以预测两种细胞型间的DGE值。dcsDiff首先利用多个自编码器(AutoEncoder, AE)和双向长短时记忆(Bi‑directional Long Short‑Term Memory, Bi‑LSTM)网络[40]减小输入规模,对bin连接信息和HM模块化特性进行建模,得到各细胞条件下各HM的嵌入表示。这些嵌入表示分别输入到多个卷积神经网络(Convolutional Neural Network, CNN)[41]中。其中:s‑CNNs分别挖掘每一类细胞型特异的HM组合效应;d‑CNN用于挖掘两类细胞型间每种HM的共性和差异性以及所有类型HM的联合影响。最后,dcsDiff融合细胞型特异性信息和细胞型间异同信息来预测两种细胞型下的DGE值。通过利用s‑CNNs和d‑CNN分别对单一细胞型特异性HM信息和两细胞型间HM异同信息有针对性地建模,dcsDiff可以更全面地获取HM信号的丰富信息。利用AE降低维数,dcsDiff可以进一步降低运行时间。此外,dcsDiff还利用单一细胞型的CS GE预测反向传播以继续优化该细胞型的特异性HM组合信息表示,进一步提高DGE的预测精度。基于这些优点,dcsDiff在REMC(Roadmap Epigenomics Mapping Consortium)数据库[42]的10种不同生物细胞型对上的预测精度均优于SVR[26]、DeepChrome[37]、AttentiveChrome[38]和DeepDiff[39]等代表性相关方法,且运行速度也比大多数方法高得多。进一步的实验也证实了dcsDiff可以准确检测出差异表达的基因。
1 相关工作
根据预测是否在单一细胞型条件下进行,GE预测方法可分为细胞型特异性基因表达(CS GE)预测和差异性基因表达(DGE)预测两大类。
CS GE预测的目的是推断单一细胞型条件下的GE。文献[34]中利用线性回归来量化来自人类T细胞数据集[35]的HM和GE的关系。文献[35]中首先把每个转录本的转录起始位点(TSS)和转录终止位点(Transcription Termination Site, TTS)两侧的DNA区域切割成大小为100个碱基对(base pair, bp)的小bin(即把DNA切割成小箱),计算每个bin在对应HM的平均HM信号值,得到每个bin的输入矩阵。然后,使用多个bin特异的支持向量机(Support Vector Machines, SVM)对每个bin建模,以预测蠕虫数据集[43]上转录本的表达。文献[36]中采用特征选择,选择与GE相关性最大的最佳bin作为输入,然后在人类数据集[44]上对HM信号利用随机森林分类器进行GE的高低分类,从而评价HM对GE的影响。文献[37]中使用CNN自动学习HM之间的组合效应来对GE进行高低分类,并使用基于类优化的技术来可视化学习模型。文献[38]中利用多个基于注意力的Bi‑LSTM模块对输入的HM信号进行编码,并对HM组合交互作用进行建模,将GE进行高低分类,以理解HM模式对GE的影响。然而,当需要一致性地分析不同细胞型中基因差异性表达时,CS GE预测方法不能捕获每个基因在不同细胞型中HM的差异模式,直接进行DGE预测是更优的选择。
DGE预测的目的是直接预测两种不同细胞型条件下的基因差异表达值。文献[26]中引入了双层SVR框架,将转录因子结合位点数据和HM数据分别在小鼠胚胎干细胞和神经前体细胞条件下的差异作为输入,预测DGE值。文献[27]中考虑了癌症基因组图谱(The Cancer Genome Atlas, TCGA)[45]中配对肺癌、相邻正常组织的CpG甲基化阵列数据,以及DNA元素百科全书(ENCyclopedia Of DNA Elements, ENCODE)[46]项目中组蛋白标记的染色质免疫共沉淀技术(CHromatin ImmunoPrecipitation, CHIP‑seq)数据,使用文献[47]中提出的ReliefF算法进行特征选择,然后使用随机森林分类来预测TCGA肺癌与正常组织的DGE值。文献[39]中基于多个Bi‑LSTM,利用细胞型特定的GE预测辅助任务来鼓励DGE预测任务进行更丰富的HM特征嵌入。这些DGE预测方法有的直接利用两种细胞型间HM信号的差异值作为输入,没有考虑到细胞型特异性HM信息[26];有的将细胞型特定的HM信号值和不同细胞型间HM信号的差异值简单拼接起来,同时探索细胞型特异性HM信息和细胞型间HM差异信息,忽略了细胞型间HM差异模式与单一细胞型CS HM模式[39]的不同。为了充分利用细胞型特异性HM信息和两种不同细胞型间的HM差异信息,有必要根据这两种信息的特点,构建更有效的模型,以不同的方式描述这两种信息。此外,早期的GE预测方法往往忽略了HM信号内各个bin之间的连接关系[26,35-36]以及HM信号的模块化特性[37],可能会导致重要表观遗传信息的丢失,而基于深度学习的方法往往缺乏对HM信号信息的精细建模[38-39]。再者,前面提到的方法大多具有维度高、计算量大的缺点[26,35,38-39]。
2 问题定义
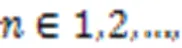
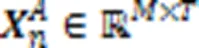
2)在细胞型和间的DGE值定义为基因在中表达值与在中表达值的对数倍变化。
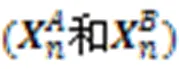
3 dcsDiff方法
dcsDiff旨在通过挖掘不同细胞型的HM信号模式,预测基因的差异表达值,寻找差异表达的基因。dcsDiff中有三个核心部分:AE‑LSTMs、s‑CNNs和d‑CNN,如图1所示。AE‑LSTMs由多个AE和Bi‑LSTM组成,它们分别对每个细胞型的每种模块化的HM信号数据进行建模得到嵌入表示,之后这些嵌入表示分别输入到d‑CNN和s‑CNNs。d‑CNN首先提取两细胞型之间每种HM的共性和细微差异,然后综合捕获所有HM的联合效应信息,获得两种细胞型对应的所有HM信号的异同信息表示。s‑CNNs挖掘每种细胞型中特异的HM组合效果,以分别获得每一细胞型的特异性表示。通过细胞型特异性的GE预测和反向传播以继续优化相应细胞型的特异性信息表示。最后,多层感知机(Multi‑Layer Perceptron, MLP)将融合前面s‑CNNs和d‑CNN挖掘出的重要的细胞型特异性信息、两细胞型间异同信息,最终预测两种细胞型间DGE值。
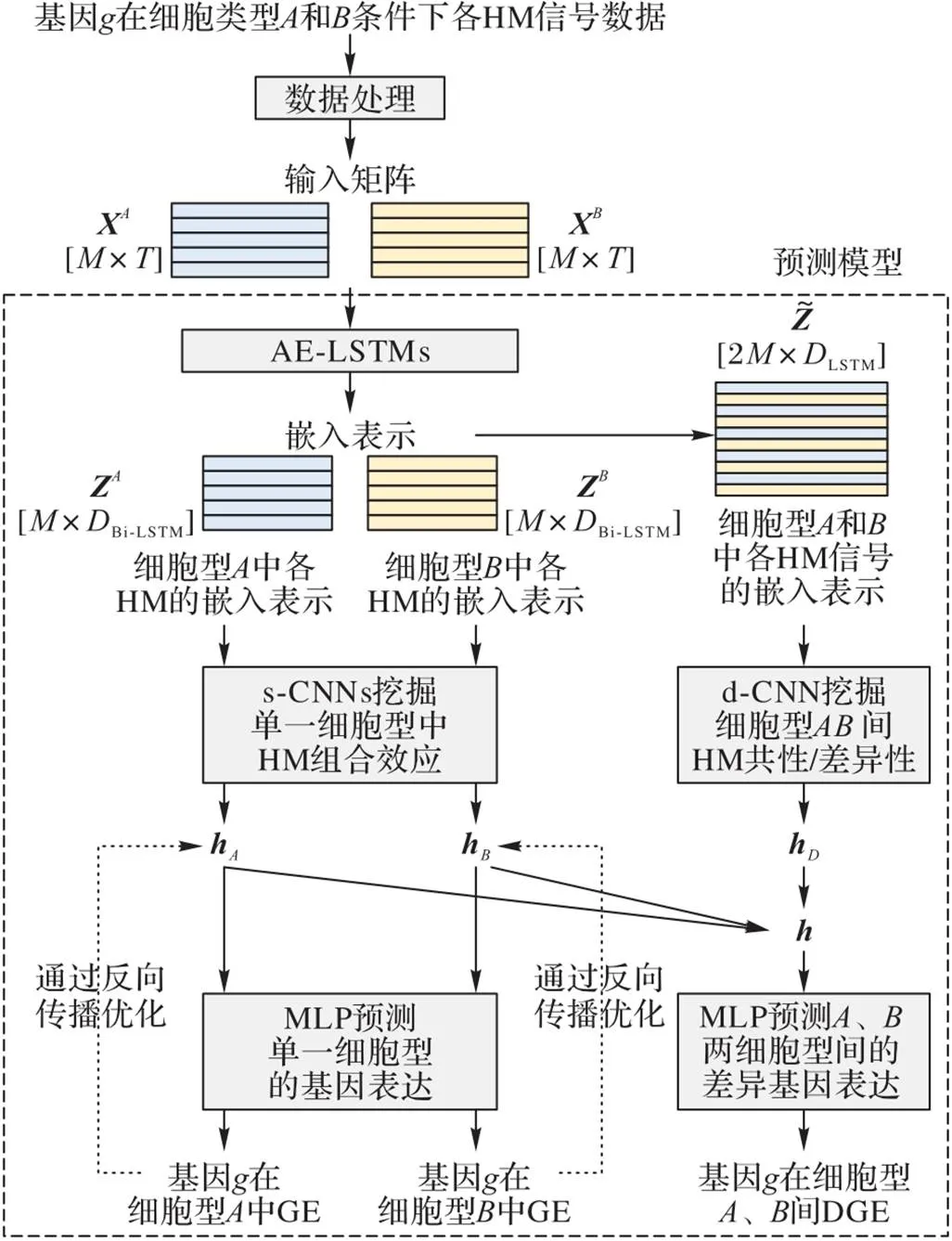
图1 dcsDiff的框架
3.1 数据预处理
首先用binning策略对HM信号数据进行处理,得到每个基因的输入矩阵和,如图2所示,数据详情见4.1.1节。
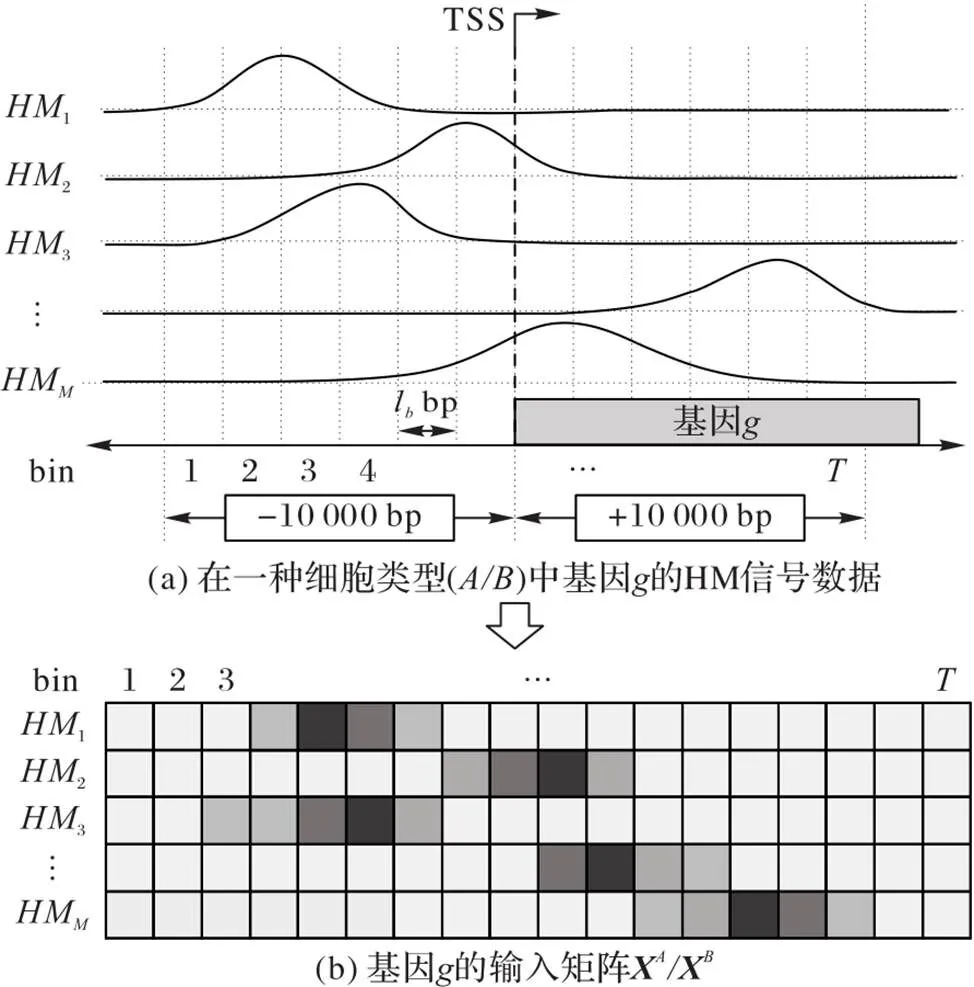
图2 每个基因在每两种细胞型下的数据处理策略
3.2 AE‑LSTMs获取HM嵌入表示
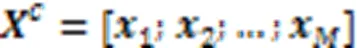
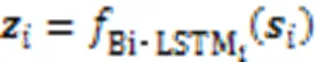
3.3 d‑CNN捕获细胞型间HM异同信息
在不同的生物组织类型或细胞发育阶段,基因的表达可能因HM谱的变化而存在差异。为了探索两种细胞型之间可能导致不同程度DGE的HM异同信息,引入了基于CNN的d‑CNN。它首先对两种细胞型之间的每类HM提取异同特征,然后捕获所有HM的异同联合效应,最后得到细胞型和的HM异同表示,具体如下。
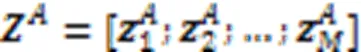
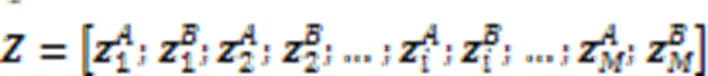
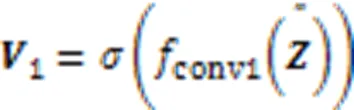
其中是激活函数整流线性单元(Rectified Linear Unit, ReLU)[48]。接下来是最大池化层,池化大小为(1,1),用于降维:
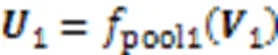
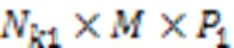
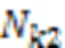
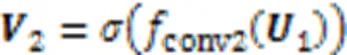
2存储了所有HM的异同组合效应。接下来是一个最大池化层,池化大小为(1,2),得到压缩的特征矩阵2:
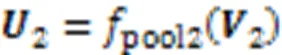
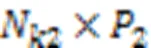
3.4 s‑CNNs捕获各细胞型特异性HM信息
每种细胞型的GE水平受该细胞型下HM的组合模式所影响。引入每个细胞型的CS HM信息可以提高DGE预测精度,然而单一细胞型的CS HM模式不同于两个细胞型间HM异同模式。因此,引入s‑CNNs对CS HM信号数据进行有针对性地建模,以充分利用每个特定细胞型的CS HM信息。s‑CNNs分别对每种细胞型的HM信号数据进行建模,利用CNN来挖掘每个特定细胞型条件下的HM组合效应,分别为细胞型和获得CS HM表示。
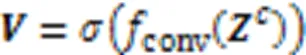
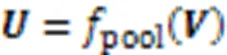
3.5 单一细胞型的CS GE预测
由于每个细胞型的特异性HM信息的引入可以提高DGE预测精度,已于3.4节获取了每个细胞型的CS HM表示。为了更准确地描述CS HM表示,本文算法还采用了基于反向传播优化的CS GE预测方法。
为了获得CS HM表示的更准确描述以用于后续DGE预测,将输入MLP进行CS GE预测:
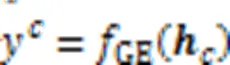
3.6 细胞型间DGE预测
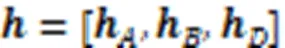
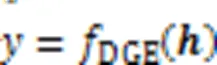
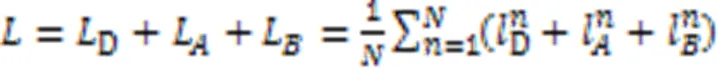
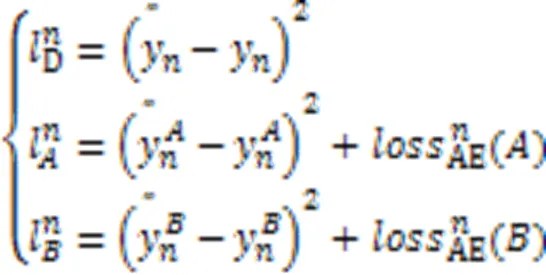
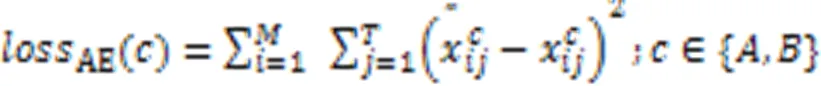
4 实验与结果分析
4.1 实验设置
4.1.1数据集
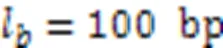

表1 组蛋白修饰及相关基因组区域
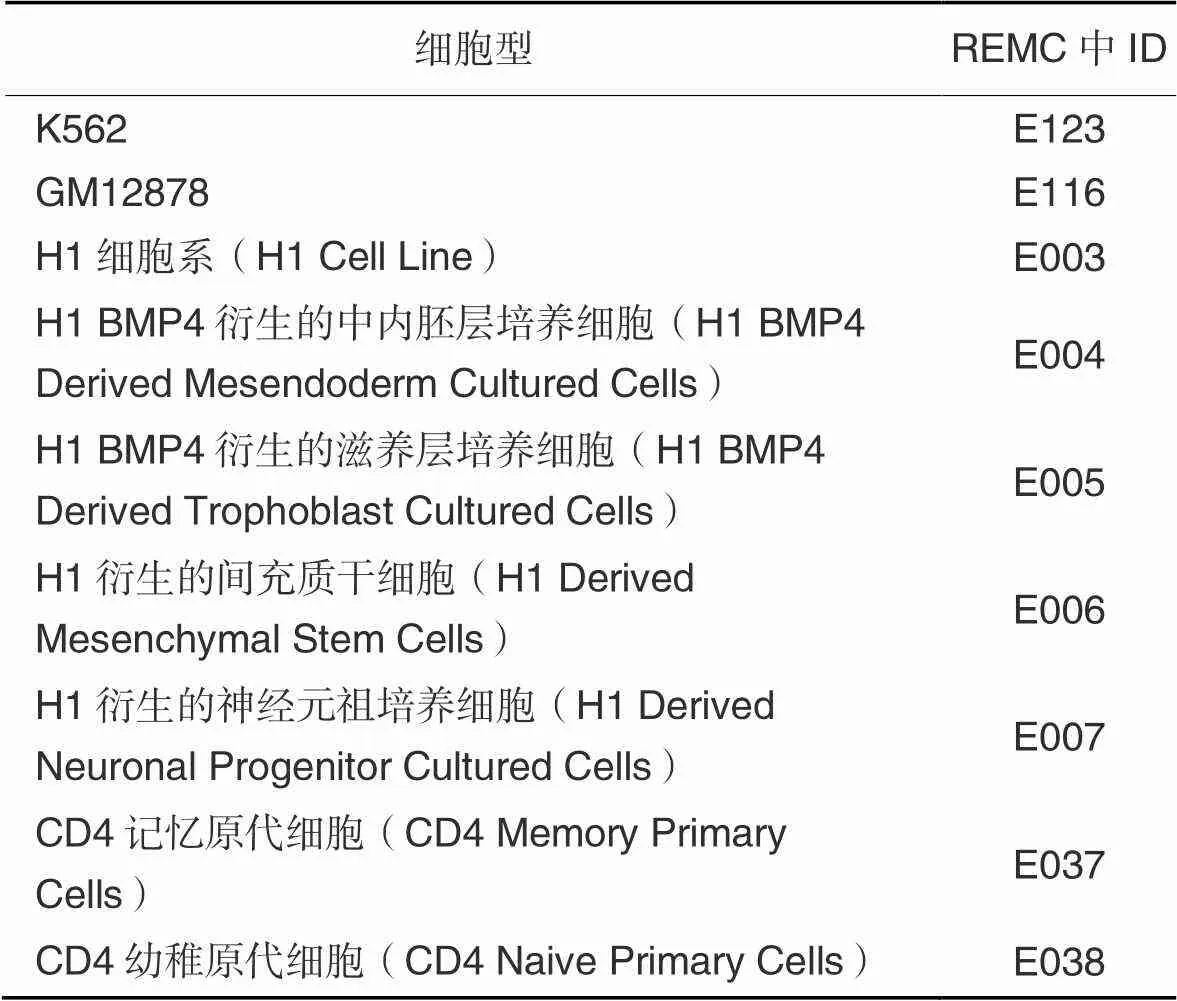
表2 REMC数据库中选择的9种细胞型和ID

表3 选择的实验细胞型对
4.1.2对比方法
将本文方法与以下几种具有代表性的相关方法进行对比:SVR[26]、DeepChrome[37]、AttentiveChrome[38]和DeepDiff[39]。因为很少有在HM层面预测DGE的方法,DeepChrome和AttentiveChrome作为先进的CS GE预测方法,也被选做对比方法,它们也可以间接用于DGE预测。SVR和DeepDiff都是DGE预测方法。
对于CS GE分类方法DeepChrome和AttentiveChrome,分别在9种细胞型中训练它们为CS GE回归任务,以预测单一细胞型下的GE值,然后计算选定的10种细胞型对之间的DGE值。所有这些对比方法都遵循作者的实验设置建议实施。用scikit‑learn[50]包重新实现SVR,用Pytorch重新实现DeepChrome和AttentivChrome,并直接采用DeepDiff的共享源代码。
4.1.3参数设置
4.1.4评价指标
选择的评价指标是皮尔逊相关系数(Pearson Correlation Coefficient, PCC),它通常用于DGE预测。PCC是两个变量之间协方差和标准差的商,定义如下:
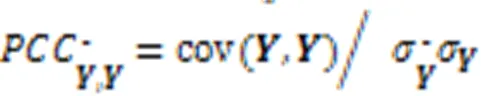
4.2 基因的差异性表达预测结果
dcsDiff和对比方法在10种细胞型对上的DGE预测结果如图3所示。在大多数细胞型对中,SVR在所有比较方法中表现最差,因为它单独建模每个bin,忽略了每个HM内部各bin的连接关系;作为一个深度学习框架,DeepChrome自动整合bin信息的局部和全局特征,在大多数细胞型对中比SVR的性能更好;AttentiveChrome分别建模每个HM以考虑它们的模块化信息,但DeepChrome直接集成所有HM导致丢失了这种信息,因此AttentiveChrome比DeepChrome表现更好;DeepDiff利用与AttentiveChrome相似的基于Bi‑LSTM的框架,将CS HM信号数据和HM信号差值数据简单拼接作为DGE预测的输入,它比AttentiveChrome的结果更好说明了DGE预测的必要性。
在细胞对E003‒E004和E037‒E038中,两种细胞型对之间的HM模式可能存在较大差异。DeepChrome和AttentiveChrome分别预测两种细胞型的CS GE,然后再计算DGE;而SVR和DeepDiff则分别将两种细胞型的差值矩阵作为输入来预测DGE。后者更偏好两种细胞型间HM差异较大的输入,因此SVR在E003‒E004和E037‒E038中的性能甚至比DeepChrome更好,且DeepDiff和AttentiveChrome在E003‒E004和E037‒E038中的性能差距比其他细胞型对中更大。在所有细胞型对中,dcsDiff优于其他所有的对比方法。dcsDiff结合CNN和Bi‑LSTM对bin的连接关系和模块化特性进行建模,避免了重要表观遗传信息丢失,从而优于SVR等传统机器学习方法。dcsDiff引入d‑CNN和s‑CNNs分别提取两种细胞型间HM异同信息和每类细胞型的特异性HM信息,从而实现了对HM模式的精确建模,比现有的深度学习方法性能更好。此外,采用自编码器降低输入数据维数,dcsDiff比大多数对比方法花费更少的运行时间(详情见第4.6节)。
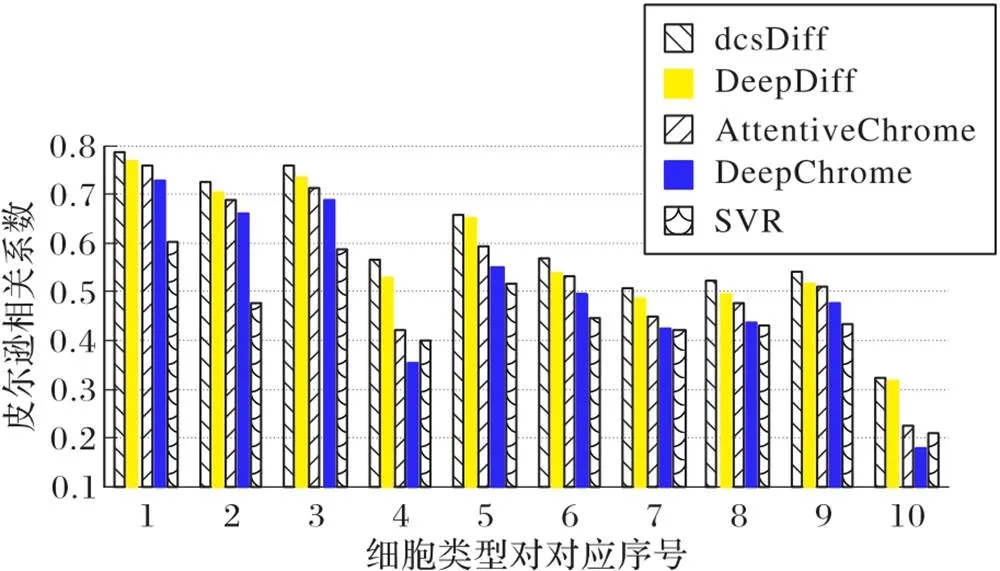
图3 各细胞型对中dcsDiff和对比方法的PCC
4.3 差异表达基因检测结果
4.4 dcsDiff成分分析
为了进一步分析dcsDiff中各成分的重要性,构建变种实验来研究它们在DGE预测中的有效性。其中:dcsDiff‑l只利用AE‑LSTMs建模bin连接信息和HM模块化特性;dcsDiff‑d包含AE‑LSTMs和d‑CNN,探索细胞型间的HM异同信息;dcsDiff‑s包含AE‑LSTMs和s‑CNNs,捕获两种细胞型各自的CS HM信息;dcsDiff结合了AE‑LSTMs、d‑CNN和s‑CNNs来整合这些信息进行DGE预测。结果如图4所示:dcsDiff‑l在所有变种中表现最差,说明仅利用每种细胞型下每种HM的bin连接信息和HM模块化特性来预测DGE是不够的。dcsDiff‑s的效果比dcsDiff‑d的效果好,表明在大部分情况下细胞型特异性HM信息比两细胞型间HM异同信息更重要。如前所述,在细胞型对E003‒E004和E037‒E038中可能存在较大的HM差异,dcsDiff‑d比dcsDiff‑s更偏好差异较大的输入,在这两个细胞型对下dcsDiff‑d和dcsDiff‑s的性能相当也证实了这一点。dcsDiff将两种细胞型间的HM异同信息和每种细胞型的特异性HM信息进行集成,总能获得最佳的性能,说明合理整合这两种信息对DGE预测是有效和必要的。
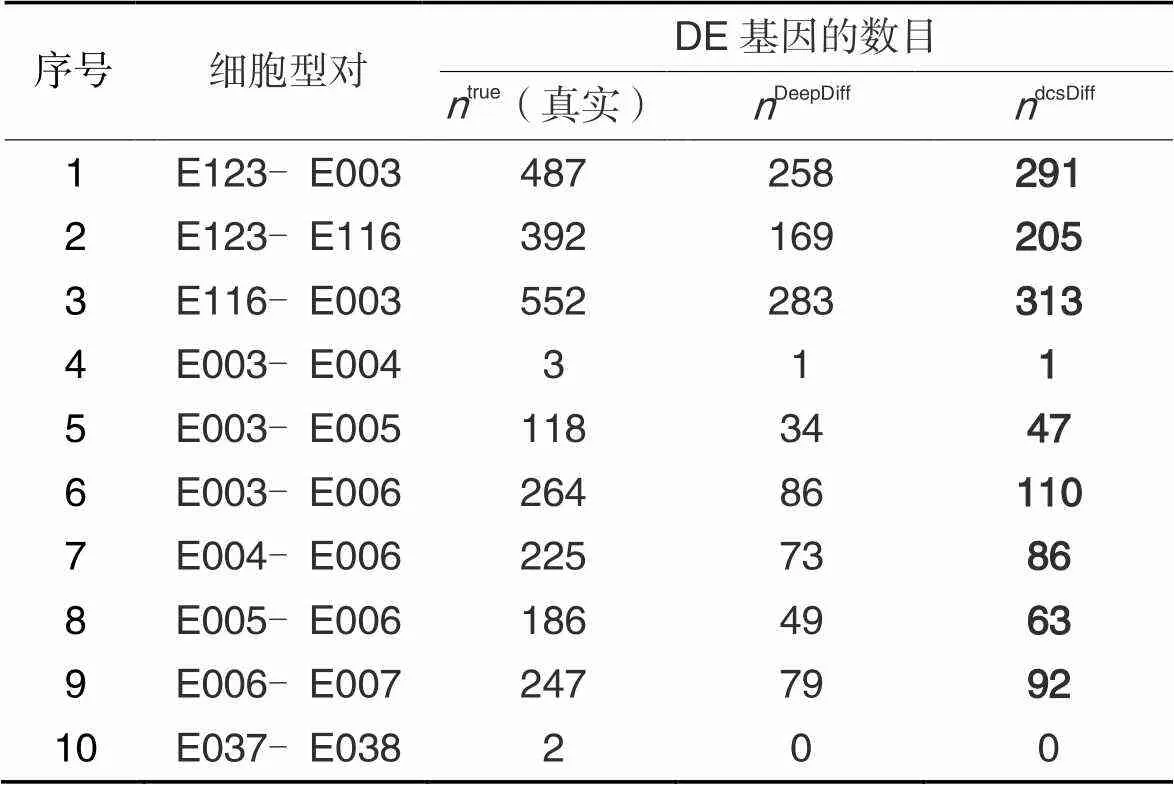
表4 每个细胞型对的DE基因以及被dcsDiff和 DeepDiff正确检测到的DE基因的统计
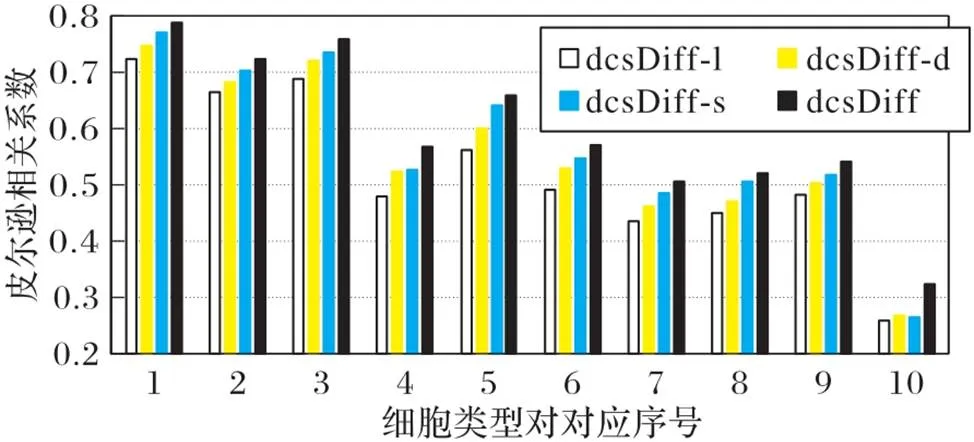
图4 10个细胞型对中不同dcsDiff变种的PCC
4.5 bin对dcsDiff的影响分析
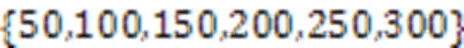
4.6 运行时间分析
本文算法在一个中等大小的服务器(Ubuntu 16.04、Intel Xeon Platinum 8163 CPU @ 2.50GHz、NVIDIA TITAN V GPU、1TB RAM)上进行实验,以研究dcsDiff与其他对比方法的运行时间。所有的对比方法都用Python 3.6实现。为了公平起见,采用相同的批大小训练了所有的深度学习方法(DeepChrome、AttentiveChrome、DeepDiff和dcsDiff)。由于scikit‑learn不支持GPU,因此并行运行SVR模型中所有的bin特异性SVR。实验结果见表6。
图5 不同输入大小下dcsDiff的PCC
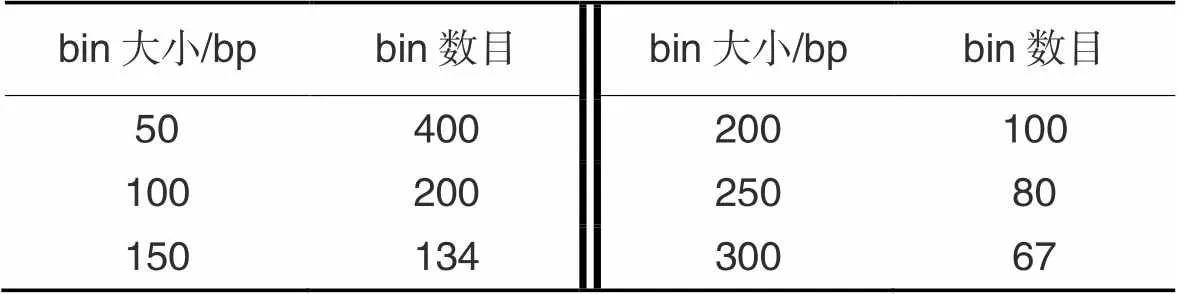
表5 bin大小及相应的bin数目
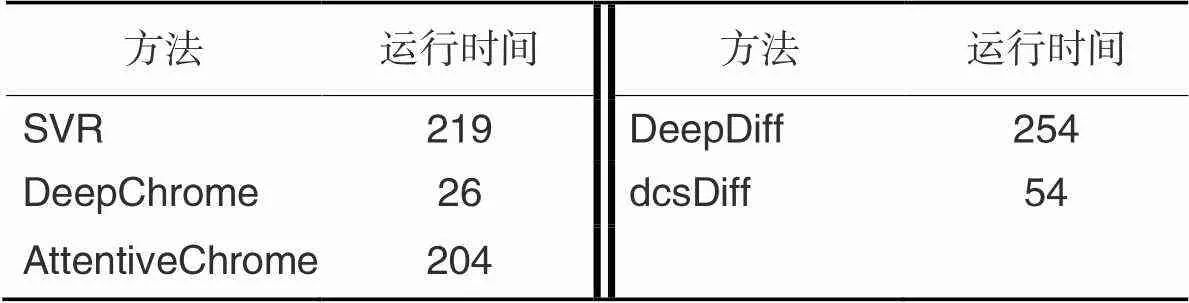
表6 运行时间统计 单位: min
由表6可知,即使是在并行计算的情况下,SVR仍然比dcsDiff有更长的运行时间,因为它利用bin特异的SVR来建模每个bin,因此在每个训练过程中需要训练太多的模型。DeepDiff和AttentiveChrome都是基于Bi‑LSTM的方法,它们利用多个Bi‑LSTM来建模输入的HM,也需要很长的运行时间,因为Bi‑LSTM需要建模长依赖特性。DeepChrome只使用一个CNN来建模所有HM信息,所以它需要最少的运行时间。尽管dcsDiff需要对两种细胞型间异同信息和细胞型特异性信息进行建模,而DeepChrome只建模一种细胞型的细胞型特异性信息,dcsDiff通过在Bi‑LSTM之前使用自编码器对输入规模进行压缩以减少运行时间,通常在30个epoch内收敛,而DeepChrome需要迭代100个epoch。综上所述,dcsDiff方法不仅比其他方法具有更高的DGE预测精度,而且运行速度也比大多数方法快得多。
5 结语
HM在GE调控中起着重要作用,它们可以协同引起DGE现象。通过大规模的HM数据预测DGE,检测不同细胞型之间的差异表达基因,可以帮助研究人员更好地理解表观遗传机制。然而,目前的解决方案往往面临各种信息利用不足、建模不精确、输入规模大、耗时长等问题。本文提出了基于Bi‑LSTM和CNN等深度学习框架的方法dcsDiff,分别对细胞型间HM异同信息和细胞型特异性HM信息进行精确建模,并最终融合这些信息来预测两种细胞型之间的差异基因表达。dcsDiff不仅优于现有的对比方法,而且运行速度也比大多数对比方法快。本文研究表明,在信息融合前对不同信息分别进行更精确的建模可以提高差异表达基因检测的准确性。基于其较高的准确性和效率,dcsDiff可作为预测差异基因表达的新框架。未来的工作将探索更多与差异基因表达相关的表观遗传修饰。
[1] BANNISTER A J, KOUZARIDES T. Regulation of chromatin by histone modifications[J]. Cell Research, 2011, 21(3): 381-395.
[2] HEINTZMAN N D, STUART R K, HON G, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome[J]. Nature Genetics, 2007, 39(3): 311-318.
[3] BONASIO R, TU S J, REINBERG D. Molecular signals of epigenetic states[J]. Science, 2010, 330(6004): 612-616.
[4] LI B, CAREY M, WORKMAN J L. The role of chromatin during transcription[J]. Cell, 2007, 128(4): 707-719.
[5] LIM P S, HARDY K, BUNTING K L, et al. Defining the chromatin signature of inducible genes in T cells[J]. Genome Biology, 2009, 10(10): No.R107.
[6] CAIN C E, BLEKHMAN R, MARIONI J C, et al. Gene expression differences among primates are associated with changes in a histone epigenetic modification[J]. Genetics, 2011, 187(4): 1225-1234.
[7] GJONESKA E, PFENNING A R, MATHYS H, et al. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease[J]. Nature, 2015, 518(7539): 365-369.
[8] WENG N P, ARAKI Y, SUBEDI K. The molecular basis of the memory T cell response: differential gene expression and its epigenetic regulation[J]. Nature Reviews Immunology, 2012, 12(4): 306-315.
[9] RINTISCH C, HEINIG M, BAUERFEIND A, et al. Natural variation of histone modification and its impact on gene expression in the rat genome[J]. Genome Research, 2014, 24(6): 942-953.
[10] 刘建敏,曹蜀炜,张萌,等. RNF2基因在喉癌组织及细胞中的表达情况研究[J]. 医学综述, 2018, 24(14): 2886-2889, 2895.(LIU J M, CAO S W, ZHANG M, et al. Study of RNF2 expression in larynocarcinoma tissue and cells[J]. Medical Recapitulate, 2018, 24(14): 2886-2889, 2895.)
[11] 岳峰,孙亮,王宽全,等. 基因表达数据的聚类分析研究进展[J]. 自动化学报, 2008, 34(2):113-120.(YUE F, SUN L, WANG K Q, et al. State‑of‑the‑art of cluster analysis of gene expression data[J]. Acta Automatica Sinica, 2008, 34(2):113-120.)
[12] 闫麒,蔺亚妮,黄先琪,等. 急性髓系白血病融合基因表达特点分析[J]. 中华血液学杂志, 2021, 42(6):480-486.(YAN Q, LIN Y N, HUANG X Q, et al. Analysis of fusion gene expression in acute myeloid leukemia[J]. Chinese Journal of Hematology, 2021, 42(6):480-486.)
[13] 雷越,万婕,文韬宇,等. siRNA沉默FOXM1基因表达对人鼻咽癌细胞增殖、凋亡及化疗敏感性的影响[J]. 肿瘤, 2018, 38(1):25-34.(LEI Y, WAN J, WEN T Y, et al. Effects of siRNA silencing the expression of FOXM1 gene on proliferation, apoptosis and chemosensitivity of human nasopharyngeal carcinoma cells[J]. Tumor, 2018, 38(1): 25-34.)
[14] 袁佳仪,何恒晶,毕娅琼,等. TOP2A基因表达对膀胱癌的预后价值分析[J]. 国际肿瘤学杂志, 2018, 45(1):22-26.(YUAN J Y, HE H J, BI Y Q, et al. Prognostic value analysis of TOP2A gene expression for bladder cancer[J]. Journal of International Oncology, 2018, 45(1): 22-26.)
[15] 刘潇,李文桂. 铜绿假单胞菌重组Bb‑OprI疫苗诱导小鼠保护力和脾细胞因子基因表达变化的研究[J]. 中国病原生物学杂志, 2018, 13(3):226-229.(LIU X, LI W G. Study on protection by and changes in expression of cytokine genes in splenocytes from mice inoculated with a recombinant Bb‑Oprl vaccine against Pseudomonas aeruginosa[J]. Journal of Pathogen Biology, 2018, 13(3):226-229.)
[16] JIA T, KULKARNI R V. Intrinsic noise in stochastic models of gene expression with molecular memory and bursting[J]. Physical Review Letters, 2011, 106(5): No.058102.
[17] QIU S G, JIA T. Quantifying the noise in bursty gene expression under regulation by small RNAs[J]. International Journal of Modern Physics C, 2019, 30(7): No.1940002.
[18] ZHANG J J, ZHOU T S. Markovian approaches to modeling intracellular reaction processes with molecular memory[J]. Proceedings of the National Academy of Sciences of the United States of America, 2019, 116(47): 23542-23550.
[19] ZHANG Z Q, DENG Q Q, WANG Z H, et al. Exact results for queuing models of stochastic transcription with memory and crosstalk[J]. Physical Review E, 2021, 103(6): No.062414.
[20] KUMAR N, JIA T, ZARRINGHALAM K, et al. Frequency modulation of stochastic gene expression bursts by strongly interacting small RNAs[J]. Physical Review E, 2016, 94(4): No.042419.
[21] OUYANG Z Q, ZHOU Q, WONG W H. ChIP‑Seq of transcription factors predicts absolute and differential gene expression in embryonic stem cells[J]. Proceedings of the National Academy of Sciences of the United States of America, 2009, 106(51): 21521-21526.
[22] XU J F, SHI J J, CUI X D, et al. Cellular Heterogeneity‑Adjusted cLonal Methylation (CHALM) improves prediction of gene expression[J]. Nature Communications, 2021, 12: No.400.
[23] NATARAJAN A, YARDIMCI G G, SHEFFIELD N C, et al. Predicting cell‑type‑specific gene expression from regions of open chromatin[J]. Genome Research, 2012, 22(9): 1711-1722.
[24] KARLIĆ R, CHUNG H R, LASSERRE J, et al. Histone modification levels are predictive for gene expression[J]. Proceedings of the National Academy of Sciences of the United States of America, 2010, 107(7): 2926-2931.
[25] HO B H, HASSEN R M K, LE N T. Combinatorial roles of DNA methylation and histone modifications on gene expression[C]// Proceedings of the 2014 National Foundation for Science and Technology Development (NAFOSTED) Conference on Information and Computer Science, AISC 341. Cham: Springer, 2015: 123-135.
[26] CHENG C, GERSTEIN M. Modeling the relative relationship of transcription factor binding and histone modifications to gene expression levels in mouse embryonic stem cells[J]. Nucleic Acids Research, 2012, 40(2): 553-568.
[27] LI Z C, GAO N, MARTINI J W R, et al. Integrating gene expression data into genomic prediction[J]. Frontiers in Genetics, 2019, 10: No.126.
[28] SCHMIDT F, KERN F, SCHULZ M H. Integrative prediction of gene expression with chromatin accessibility and conformation data[J]. Epigenetics and Chromatin, 2020, 13: No.4.
[29] AVSEC Ž, AGARWAL V, VISENTIN D, et al. Effective gene expression prediction from sequence by integrating long‑range interactions[J]. Nature Methods, 2021, 18(10):1196-1203.
[30] LI J, CHING T, HUANG S J, et al. Using epigenomics data to predict gene expression in lung cancer[J]. BMC Bioinformatics, 2015, 16(S5): No.S10.
[31] FRASCA M, PAVESI G. A neural network based algorithm for gene expression prediction from chromatin structure[C]// Proceedings of the 2013 International Joint Conference on Neural Networks. Piscataway: IEEE, 2013: 1-8.
[32] KUMAR V, MURATANI M, RAYAN N A, et al. Uniform, optimal signal processing of mapped deep sequencing data[J]. Nature Biotechnology, 2013, 31(7): 615-622.
[33] ERNST J, KELLIS M. Large‑scale imputation of epigenomic datasets for systematic annotation of diverse human tissues[J]. Nature Biotechnology, 2015, 33(4): 364-367.
[34] COSTA I G, ROIDER H G, DO REGO T G, et al. Predicting gene expression in t cell differentiation from histone modifications and transcription factor binding affinities by linear mixture models[J]. BMC Bioinformatics, 2011, 12(S1): No.S29.
[35] CHENG C, YAN K K, YIP K Y, et al. A statistical framework for modeling gene expression using chromatin features and application to modENCODE datasets[J]. Genome Biology, 2011, 12(2): No.R15.
[36] DONG X J, GREVEN M C, KUNDAJE A, et al. Modeling gene expression using chromatin features in various cellular contexts[J]. Genome Biology, 2012, 13(9): No.R53.
[37] SINGH R, LANCHANTIN J, ROBINS G, et al. DeepChrome: deep‑learning for predicting gene expression from histone modifications[J]. Bioinformatics, 2016, 32(17): i639-i648.
[38] SINGH R, LANCHANTIN J, SEKHON A, et al. Attend and predict: understanding gene regulation by selective attention on chromatin[J]. Advances in Neural Information Processing Systems, 2017, 30: 6785-6795.
[39] SEKHON A, SINGH R, QI Y J. DeepDiff: DEEP‑learning for predicting DIFFerential gene expression from histone modifications[J]. Bioinformatics, 2018, 34(17): i891-i900.
[40] HOCHREITER S, SCHMIDHUBER J. Long short‑term memory[J]. Neural Computation, 1997, 9(8): 1735-1780.
[41] LeCUN Y, BOTTOU L, BENGIO Y, et al. Gradient‑based learning applied to document recognition[J]. Proceedings of the IEEE, 1998, 86(11): 2278-2324.
[42] Roadmap Epigenomics Consortium, KUNDAJE A, MEULEMAN W, et al. Integrative analysis of 111 reference human epigenomes[J]. Nature, 2015, 518(7539): 317-330.
[43] CELNIKER S E, DILLON L A L, GERSTEIN M B, et al. Unlocking the secrets of the genome[J]. Nature, 2009, 459(7249): 927-930.
[44] The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome[J]. Nature, 2012, 489(7414): 57-74.
[45] TOMCZAK K, CZERWIŃSKA P, WIZNEROWICZ M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge[J]. Contemporary Oncology/Współczesna Onkologia, 2015, 19(1A): A68-A77.
[46] On behalf of The ENCODE Project Consortium. The ENCODE (ENCyclopedia of DNA elements) project[J]. Science, 2004, 306(5696): 636-640
[47] KONONENKO I, ŠIMEC E, ROBNIK‑ŠIKONJA M. Overcoming the myopia of inductive learning algorithms with RELIEFF[J]. Applied Intelligence, 1997, 7(1): 39-55.
[48] NAIR V, HINTON G E. Rectified linear units improve restricted Boltzmann machines[C]// Proceedings of the 27th International Conference on Machine Learning. Madison, WI: Omnipress, 2010: 807-814.
[49] KINGMA D P, BA J L. Adam: a method for stochastic optimization[EB/OL]. (2017-01-30)[2021-06-30].https://arxiv.org/pdf/1412.6980.pdf.
[50] PEDREGOSA F, VAROQUAUX G, GRAMFORT A, et al. Scikit‑learn: machine learning in Python[J]. Journal of Machine Learning Research, 2011, 12: 2825-2830.
Deep fusion model for predicting differential gene expression by histone modification data
LI Xin, JIA Tao*
(,,400715,)
Concering the problem that the Cell type‑Specificity (CS) and similarity and difference information between different cell types are not properly used when predicting Differential Gene Expression (DGE) with large‑scale Histone Modification (HM) data, as well as large volume of input and high computational cost, a deep learning‑based method named dcsDiff was proposed. Firstly, multiple AutoEncoders (AEs) and Bi‑directional Long Short‑Term Memory (Bi‑LSTM) networks were introduced to reduce the dimensionality of HM signals and model them to obtain the embedded representation. Then, multiple Convolutional Neural Networks (CNNs) were used to mine the HM combined effects in each single cell type, and the similarity and difference information of each HM and joint effects of all HMs between two cell types. Finally, the two kinds of information were fused to predict DGE between two cell types. In the comparison experiments with DeepDiff on 10 pairs of cell types in the REMC (Roadmap Epigenomics Mapping Consortium) database, the Pearson Correlation Coefficient (PCC) of dcsDiff in DGE prediction was increased by 7.2% at the highest and 3.9% on average, the number of differentially expressed genes accurately detected by dcsDiff was increased by 36 at most and 17.6 on average, and the running time of dcsDiff was saved by 78.7%. The validity of reasonable integration of the above two kinds of information was proved in the component analysis experiment. The parameters of dcsDiff were also determined by experiments. Experimental results show that the proposed dcsDiff can effectively improve the efficiency of DGE prediction.
Histone Modification (HM); Differential Gene Expression (DGE); Cell type‑Specificity (CS); AutoEncoder (AE); Bi‑directional Long Short‑Term Memory (Bi‑LSTM) network; information fusion; epigenetics
This work is partially supported by Industry‑University‑Research Innovation Fund for Universities of China, Ministry of Education (2021ALA03016).
LI Xin, born in 1997, M. S. candidate. Her research interests include data mining, bioinformatics, machine learning.
JIA Tao, born in 1982, Ph. D., professor. His research interests include data science, complex network.
1001-9081(2022)11-3404-09
10.11772/j.issn.1001-9081.2021111956
2021⁃11⁃17;
2021⁃11⁃23;
2021⁃12⁃06。
教育部中国高校产学研创新基金资助项目(2021ALA03016)。
TP301.6
A
李昕(1997—),女,四川绵阳人,硕士研究生,CCF会员,主要研究方向:数据挖掘、生物信息学、机器学习;贾韬(1982—),男,重庆人,教授,博士,CCF会员,主要研究方向:数据科学、复杂网络。
