自噬调节脓毒症细胞焦亡机制的研究进展
2022-11-24王雅琪温继梨
王雅琪 温继梨
1.内蒙古医科大学第一临床医学院,内蒙古呼和浩特 010000;2.内蒙古医科大学附属医院急诊内科,内蒙古呼和浩特 010000
脓毒症(sepsis)是由感染引起的全身炎症反应综合征,每年估计有4890 万起因脓毒症和1100 万与脓毒症相关的死亡[1-3]。目前脓毒症的治疗多采用广谱抗生素、源头控制和支持性治疗,这些非特异性治疗并不能降低脓毒症的高病死率。脓毒症已然成为全球公共卫生的一大挑战,需要科研人员深入研究脓毒症的病理生理机制,寻求新的诊治靶点。
焦亡是由gasdermin D(GSDMD)介导的溶解性细胞死亡的促炎症模式,是最具特征性的反应[4]。炎性小体激活介导促炎细胞因子白细胞介素(interleukin,IL)–1b 和IL–18 的蛋白水解加工及胱天蛋白酶分泌,从而诱导裂解型细胞死亡。焦亡首先在巨噬细胞及其相关疾病中被发现[5],随后大量报道证实中性粒细胞也可发生焦亡,感染期间IL–18主要由中性粒细胞和单核细胞分泌[4]。血清IL–18 水平升高与脓毒症的严重程度和预后相关[6,7]。研究证实过度激活焦亡会导致器官损伤,抑制焦亡可能是脓毒症治疗的新方向。
自噬是细胞生存的重要自我保护机制,通过多种途径在脓毒症中起保护作用,包括清除病原体、抑制炎性反应、预防免疫抑制和调节代谢。近年来研究已经证实自噬对细胞焦亡有调节作用,本文重点阐述有关自噬对脓毒症细胞焦亡调节作用的最新研究成果。
1 细胞焦亡的新进展
焦亡主要发生在巨噬细胞及其前体、胱天蛋白酶–1 和胱天蛋白酶–11 激活后的单核细胞中。炎性小体激活巨噬细胞中被胱天蛋白酶–1 切割GSDMD 产生的N–GSDMD 片段,随后N–GSDMD 片段在质膜中寡聚化,形成增加质膜通透性的孔,导致细胞焦亡和IL–1β 释放[8,9]。Karmakar 等[10]发现中性粒细胞与巨噬细胞运输GSDMD 存在根本性差异,中性粒细胞中的N–GSDMD 片段与嗜天青颗粒和LC3+自噬体相关,IL–1β 则是通过自噬依赖性机制分泌,这些差异是炎性小体激活过程中中性粒细胞特异性功能的基础。细胞溶质脂多糖(lipopolysaccharide,LPS)激活胱天蛋白酶–11 依赖性非经典炎性小体的能力受到多种机制调控,以避免过度炎性反应。最新研究证明L–肾上腺素不仅可作用于α–(2B)肾上腺素能受体抑制胱天蛋白酶–11 的激活,还可通过腺苷酸环化酶4 诱导环磷酸腺苷生成促进蛋白激酶A活化,从而阻断胱天蛋白酶–11 介导的IL–1 分泌、GSDMD 裂解及随后的损伤相关分子模式(damage associated molecular pattern,DAMP)激活[11]。这可能是免疫代谢作为脓毒症潜在治疗靶点的概念性证明。
脓毒症引起的休克和组织损伤需要胱天蛋白酶–11 激活、切割GSDMD、受体相互作用蛋白激酶3(receptor–interacting protein kinase–3,RIPK3)和混合谱系激酶结构域样蛋白磷酸化。Chen 等[12]研究表明,细胞焦亡中RIPK3 和GSDMD 具有协同作用,可放大巨噬细胞和内皮细胞的坏死和组织因子释放,从而导致组织损失。临床数据分析低镁血症与脓毒症患者的单核细胞计数减少有关,镁离子通过限制N–GSDMD 末端激活时的寡聚化和膜定位抑制细胞焦亡[13]。焦亡发生在巨噬细胞中已广为人知,但多形核中性粒细胞衍生的外泌体在脓毒症中的潜在作用机制尚不清楚,Jiao 等[14]通过动物实验发现,中性粒细胞的外泌体miR–30d–5p 通过激活核因子κB(nuclear factor–κB,NF–κB)M1 型巨噬细胞极化引发巨噬细胞焦亡。
2 自噬调节脓毒症细胞焦亡
2.1 分子机制
焦亡依据不同的起始因子和有效分子分为经典途径和非经典途径,见图1。自噬通过消除DAMP和病原体相关分子模式(pathogen associated molecular pattern,PAMP)下调焦亡。DAMP 被定义为能够启动和增强炎性反应的内源性因子,PAMP则是指外源性微生物产物,如LPS[15,16]。DAMP 的自噬消除与线粒体功能受损有关。研究发现自噬通过下调裂解的GSDMD 水平来防止焦亡[17]。作为自噬激动剂的雷帕霉素可逆转LPS 刺激后GSDMD 介导的焦亡,但单独对焦亡没有抑制作用,强调自噬在整个过程中的重要性[18]。既往研究提到自噬对GSDMD 介导的焦亡也具有调节作用,可能与AMPK–eEF–2K 信号通路有关[19,20],但具体机制仍不清楚,需进一步深入研究。
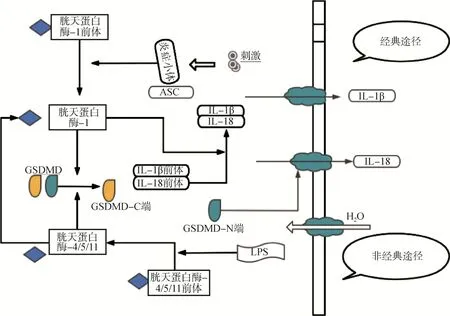
图1 经典和非经典细胞焦亡途径
2.2 自噬、焦亡和一些经典的信号通路
有多个信号通路参与自噬的调节,但确切的作用机制尚未阐明。过氧化物酶体增殖物激活受体γ(peroxisome proliferator activated receptor γ,PPARγ)具有抗炎抗氧化的作用,对自噬和焦亡均有调控作用,PPARγ 表达增加、活性氧水平降低可抑制硫氧还蛋白互作蛋白(thioredoxin interacting protein,TXNIP)/核苷酸结合寡聚化结构域样受体蛋白(nucleotide–binding oligomerization domain–like receptor protein 3,NLRP3)信号通路,减少脓毒症期间细胞焦亡和减轻肝功能障碍[21]。消退素D2(resolvin D2,RvD2)是由ω3–多不饱和脂肪酸产生的炎症先天抑制因子,研究已证实RvD2 对多种炎症信号通路有调节作用[22,23]。最新研究报道RvD2 可通过自噬促进NLRP3 炎性小体降解,但其潜在的机制仍需进一步研究以明确。另有研究表明miR–30c–5p通过TXNIP/NLRP3 信号通路对焦亡和脓毒症诱导的损伤进行负面控制,这可能是脓毒症急性肾损伤患者的治疗靶点[24]。
IL–17 信号通路的上、下游分子主要包括高迁移率族蛋白B1、晚期糖基化终末产物的多配体受体、IL–17A、肿瘤坏死因子受体相关蛋白和NK–κB,研究人员通过生物学信息分析发现NK–κB 的激活促进依赖胱天蛋白酶–1 的炎性反应发展,提示IL–17信号通路与肺炎导致的脓毒症显著相关[25],但还需要进一步验证。Ke 等[26]在体外建立脓毒症小鼠模型,评估IL–33 对NF–κB/P38 促分裂原活化蛋白激酶(mitogen–activated protein kinase,MAPK)信号通路和巨噬细胞焦亡的影响,结果表明IL–33 激活NF–κB/P38MAPK 信号通路,上调胱天蛋白酶–1 前体表达,从而介导脓毒症小鼠巨噬细胞焦亡。提示IL–33 可增加脓毒症小鼠巨噬细胞的焦亡水平,为临床脓毒症患者的诊断和治疗提供理论依据。脓毒症是急性肾损伤的常见危险因素,骨髓间充质干细胞 具有多向分化潜能,有研究探讨骨髓间充质干细胞在脓毒症诱导的急性肾损伤中的作用,结果表明骨髓间充质干细胞可通过上调SIRT1/Parkin 抑制炎性反应并促进肾小管上皮细胞和小鼠模型细胞的线粒体自噬,从而抑制细胞焦亡,改善脓毒症引起的肾脏损伤[27]。
干扰素基因刺激物也是脓毒症的重要因素,与不受控制的细胞焦亡相关,胞质DNA 通过GMP–cGAMP 途径激活干扰素基因刺激物,并通过经典和非经典途径触发细胞焦亡[28]。在研究长链非编码RNA ZFAS1 在脓毒症诱导的心功能障碍的调控网络中发现,转录因子SP1 诱导的ZFAS1 通过下调miR–590–3p 调节AMP 活化蛋白激酶(AMP–activated protein kinase,AMPK)–哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)信号通路,影响NLRP3 介导的心肌细胞自噬和焦亡,从而加重脓毒症诱导的心功能障碍[29]。这种新的ZFAS1/miR–590–3p/AMPK–mTOR 调节网络可能为开发治疗脓毒症诱导的心功能障碍提供新的治疗靶点。SESN2是一种高度保守的蛋白质,已被证实可维持内部环境稳态,研究发现SESN2 能够通过抑制内质网应激相关的NLRP3 降低GSDMD 依赖性焦亡;SESN2缺乏时可通过PERK–ATF4–CHOP 信号通路诱导NLRP3/ASC/CASP–1 依赖性细胞焦亡和促炎细胞因子产生,导致脓毒症小鼠死亡率增加[30]。
如前所述,大量研究结果支持自噬负调控焦亡,但也有一些研究得出相反的结论,认为自噬对焦亡有促进作用。在一项对骨髓源性巨噬细胞的研究中,饥饿增强自噬,但焦亡水平并没有相应降低,相反,炎性小体活性和IL–18、IL–1β 表达均上调。出现这种差异的原因尚不明确,可能是不同的病理条件和细胞类型影响自噬调节焦亡的方向。在未来的研究中,可关注自噬抑制炎性小体激活和减少焦亡的时间,以便将自噬诱导作为脓毒症患者的治疗方法。
3 自噬调节脓毒症——新的治疗靶点
有研究证明雷帕霉素可通过激活自噬,减轻肺部炎症,抑制细胞焦亡。Wang 等[31]利用盲肠结扎穿刺构建脓毒症模型,发现使用雷帕霉素治疗可保护小鼠免于感染性死亡。因此,调节自噬是脓毒症治疗的一个有效靶点。脓毒症心肌病是脓毒症常见的一个并发症,研究证实木犀草素可通过AMPK 信号传导减少细胞焦亡和增强自噬来保护心肌,其对心肌的保护作用与抗氧化、抗炎的药理活性相关。许多能够改善自噬的天然化学单体药物已被发现,如米诺环素、青藤碱、京尼平、蒲公英甾醇、卡达莫素及青蒿琥酯,它们均是改善脓毒症器官功能障碍的潜在治疗药物。
目前已开发出以胱天蛋白酶、GSDMD 蛋白为靶点的新药。脓毒症相关脑病临床表现为急性或长期认知功能障碍,胱天蛋白酶–1 抑制剂VX765 可有效逆转盲肠结扎穿刺诱导的脓毒症小鼠的认知功能障碍;VX765 抑制胱天蛋白酶–1 后,GSDMD 及其裂解产物GSDMD–NT 的表达减少,从而减少脑内焦亡的发生[32]。Ge 等[33]发现α–倒捻子素诱导的自噬可抑制LPS 刺激的巨噬细胞NLRP3 转录体激活,减少IL–1β 释放,继而增强巨噬细胞的吞噬功能,恢复器官功能。二甲双胍是一种经典的降糖药物,现证明对脓毒症患者具有保护作用,入院前使用二甲双胍可显著降低脓毒症患者的病死率。在许多病理情况下,如缺血再灌注损伤、炎症性肠病和糖尿病性心肌病,二甲双胍均可改善细胞焦亡。一个可能机制是二甲双胍通过增强自噬,抑制炎性小体NLRP3 的表达,减少细胞焦亡;另有研究显示AMPK–Akt–mTOR信号通路在该过程中也至关重要,但具体的机制尚不清楚。通过诱导自噬调节细胞焦亡治疗败血症的药物仍处于临床前阶段,还需更多的研究来探索自噬调节脓毒症中细胞焦亡的潜在机制。
4 小结与展望
自噬调节脓毒症中的细胞焦亡是一个较新的研究领域,其调节作用机制尚不清楚。目前已证实自噬对焦亡具有双向调节作用,机制较为复杂,涉及多种分子和物质介导的多条信号通路,这可能是未来脓毒症治疗的新线索。但如何平衡好脓毒症不同时期自噬对焦亡的影响,达到最佳的治疗效果,值得更进一步的研究与探索。
