TMEM163变异致髓鞘形成低下患者随访两例及iPSC构建*
2022-11-22张钰王君宇段若愚肖江喜吴晔姜玉武延会芳王静敏
张钰王君宇段若愚肖江喜吴晔姜玉武延会芳**王静敏
(1)北京大学第一医院儿科,北京 100034;2)儿科遗传性疾病分子诊断与研究北京市重点实验室,北京 100009;3)首都医科大学附属北京儿童医院神经内科,北京 100045;4)北京大学第一医院医学影像科,北京 100034;5)国家卫生健康委员会神经科学重点实验室,北京 100034)
髓鞘形成低下性脑白质营养不良(hypomyelinating leukodystrophy,HLD)是一类婴幼儿期起病、以发育迟缓与脑白质髓鞘形成低下为特征的神经系统遗传病。其临床及遗传学异质性强,致病机制复杂,由少突胶质细胞分化及功能异常导致,尚无治愈方法。1987年Cremers等[1]发现HLD1型(佩梅病)致病基因PLP1。至今,国际上共报道24个HLD相关致病基因,在线人类孟德尔遗传数据库(Online Mendelian Inheritance in Man,OMIM)命名为HLD1~24型。2022年,本课题组[2]主导定位TMEM163为HLD新致病基因并报道2例患儿。目前,国内外尚缺乏对TMEM163突变患者自然病程的认识,且尚无疾病模型开展机制研究。本研究现对本课题组之前报道的两例TMEM163突变患者进行随访,明确其临床及遗传学特点,并构建患者来源诱导多能干细胞(induced pluripotent stem cell,iPSC),为后续致病机制研究及药物靶点筛选打下基础。
1 对象与方法
1.1 对象
2例TMEM163变异致病患儿通过门诊、电话或微信随访获得资料,末次随访时间为2022年8月。随访内容包含临床症状、体征、血生化检查、头颅磁共振成像(MRI)、发育评估、康复训练情况等。发育评估采用Gesell发育评估量表及粗大运动功能分级(Gross motor function classification system,GMFCS)[3]进行。
病例1(Patient 1,Pt1),男,11岁。出生后至7岁发育迟缓,缓慢进步,现无临床症状。出生后发现水平性眼震,8月龄消失。运动发育轻度迟缓,6月会抬头,7月会翻身,1岁可独坐,2岁会独走。语言发育迟缓,1岁可说单词。随访发现,患儿发育缓慢进步,7岁仅表现为步态异常,进入普通小学就读。现成绩中等,步态恢复正常。无抽搐。第一胎,第一产。出生体重4 200 g。父母非近亲婚配,否认家族类似疾病史。查体:7月龄肌张力降低。7月龄头颅核磁显示脑白质髓鞘形成低下。血常规及血尿代谢筛查未见异常。2岁、7岁GMFCS评级均为I级。
病例2(Patient 2,Pt2),女,5岁1月龄。发育迟缓,缓慢进步。2月龄发现水平性眼震,2岁消失。运动发育中度迟缓,3月会抬头,10月龄会翻身,14月龄会独坐,2岁10月龄可独走伴异常步态,4岁可双腿跳跃,现可独自上楼梯。语言发育迟缓,2岁可说单词,3岁3月龄可说完整句子。现发音欠清晰,语速慢,认知社交能力正常。无抽搐。第一胎,第一产,出生体重3 489 g。父母非近亲婚配,胞兄体健,否认家族类似疾病史。查体:4月龄肌张力低,头围位于第50%百分位;15月龄肌张力低,身体生长指标、肌力、听力及视力均正常。13月龄Gesell发育评估提示运动发育中度迟缓,精细运动、语言、个人和社会适应轻度迟缓。4月龄及13月龄头颅核磁显示弥漫性髓鞘发育迟缓。肝功能、肾功能、电解质、血乳酸、同型半胱氨酸、维生素B12、先天性代谢病及溶酶体活性筛查均未见异常。2岁GMFCS评级为II级,5岁GMFCS评级为I级。
本研究经北京大学第一医院伦理委员会批准(批准号:2005-04),已获患者知情同意。
1.2 方法
1.2.1遗传学分析
软件预测利用REVEL等数据库/软件进行。蛋白质结构数据分析使用蛋白质数据库[4](UniProtKB,https://www.uniprot.org)查询蛋白质条目,利用AlphaFold[5]蛋白质模型数据库预测蛋白质结构,下载蛋白质结构文件(PDB格式),再利用VENUS[6]及DynaMut2[7]等软件,定量预测突变型和野生型蛋白质热力学稳定性差异。阈值定义为:与野生型相比,突变体结构的自由能变化(ΔΔG值)>3 kcal/mol为严重不稳定,1~3 kcal/mol为不稳定,<1 kcal/mol为中性或良性[8-9],评估变异对蛋白质结构的影响。在ClinVar、OMIM、Mastermind、PubMed、中国知网、万方数据库中进行文献调研。结合软件预测结果、蛋白质结构数据分析结果及文献内功能实验结果,参考美国医学遗传学和基因组学会(American College of Medical Genetics and Genomics,ACMG)2015年发布的变异致病性注释指南对位点进行致病性评级[10],评估变异位点致病性。
1.2.2患者来源iPSC构建
采集Pt2外周血5 ml,分离外周血单个核细胞(peripheral blood mononuclear cell,PBMC),配置PBMC培 养 基,即RPMI-1640(Thermo Fisher Scientific)/10%胎 牛 血 清(fetal bovine serum,FBS,Thermo Fisher Scientific)添加100µg/L促血小板生成素(thrombopoietin,TPO,Peprotech)、100 µg/L干 细 胞 因 子(stem cell factor,SCF,Peprotech)、100 µg/L Fms相关酪氨酸激酶3配体(Fms-related tyrosine kinase 3 ligand,FLT-3,Peprotech)、10µg/L粒 细 胞 集 落 刺 激 因 子(granulocyte colony-stimulating factor,G-CSF,Peprotech)、10µg/L IL-3(Peprotech)及GlutaMAX(Thermo Fisher Scientific)、青霉素链霉素(penicillin streptomycin,P/S,Thermo Fisher Scientific)。分离后的PBMC以1×106/ml密度用PBMC培养基培养于超低黏附6孔板的一个孔中,于5%CO2的37℃恒温培养箱中培养,每2 d换液,4 d后电转。每1×106PBMC电转10 μg非整合型诱导质粒(包含4 μg pEV-SFFV-OCT4-E2A-SOX2、4 μg pEV-SFFV-MYC-E2A-KLF4和2 μg pEV-SFFVBCL-XL)(上海捷易生物科技有限公司),电转采用Amaxa Nucleofector II电转仪的U-008程序和Amaxa® Human CD34+Cell Nucleofector® Kit(LONZA)试剂盒进行。电转后的PBMC用2 ml PBMC培养基培养于6孔板的一个孔中;铺饲养层细胞(MEF)于6孔板,以备第2天接种。配置hiPSC诱导培养基,即DMEM/F12(Thermo Fisher Scientific)添 加20% KSR(KnockOut™Serum Replacement,Thermo Fisher Scientific)、GlutaMAX、NEAA(Thermo Fisher Scientific)、P/S、0.1 mmol/L 2-巯基乙醇(2-mercaptoethanol,Thermo Fisher Scientific)、20µg/L FGF2(Fibroblast Growth Factor-basic,成纤维细胞生长因子,Peprotech)。电转后第2天,离心收取电转的PBMC,用1 ml PBMC培养基+1 ml hiPSC诱导培养基种于MEF细胞上。随后,每2 d换2 ml hiPSC诱导培养基。在电转后第10天看到小克隆出现。继续保持每2 d换液,待克隆团长到合适大小,用 枪 头 挑 取 单 克 隆,用mTeSR1(STEMCELL)种于用基质胶(matrigel,BD,#354277)包埋后的12孔板中,建系传代培养。传代第5代之后,进行鉴定。
1.2.3iPSC鉴定
a.碱性磷酸酯酶染色
碱性磷酸酶(AP)染色采用BCIP/NBT碱性磷酸酯酶显色试剂盒(碧云天)。将第10代iPSC吸弃培养基,加入PBS清洗2次,每次5 min;加入4%多聚甲醛(Sigma),室温放置20 min固定;PBS洗3次,每次5 min。按照如下比值配置BCIP/NBT染色工作液:BCIP/NBT染色工作液3.03 ml=碱性磷酸酶显色缓冲液3 ml+BCIP溶液(300×)10µl+NBT溶液(150×)20µl。将BCIP/NBT染色工作液加入培养皿中,室温避光孵育30 min,显色至合适深浅。去除工作液,用蒸馏水洗涤2次,终止显色反应。光镜下观察结果。
b.变异位点Sanger测序
利用基因组DNA提取试剂盒(宁波有成)提取患儿第5代iPSC基因组DNA,PCR扩增产物送检进行Sanger测序验证iPSC是否携带TMEM163c.227T>C变异。
c.染色体核型分析
染色体G带分析实验在北京大学细胞遗传学实验室中完成。
d.细胞免疫荧光检测
第10代的诱导多潜能干细胞用PBS清洗3次,4%多聚甲醛溶液室温固定20 min,PBS清洗3次,每 次5 min。0.2% TritonX-100(Sigma)破 膜30 min,PBS清洗3次,2%BSA封闭30 min,PBS清洗3次。加入2%BSA稀释的一抗4℃孵育过夜,次日用PBS清洗3次,与相应的二抗室温避光孵育2 h,PBS清 洗3次,每次5 min。Hoechst33342(1∶500)染细胞核,荧光显微镜(莱卡,DMi8)观察细胞并拍照。用于免疫荧光检测的抗体如下:rabbit-anti-NANOG(Abcam,#ab80892),mouseanti-OCT4(Santa Cruz,#sc-5279),goat-anti-SOX2(R&D,#AF2018),mouse-anti-TRA-1-60(Millipore,#MAB4360),donkey anti-mouse IgG 488 conjugate(Thermo Fisher Scientific,#A-21202),donkey anti-goat IgG 594 conjugate(Thermo Fisher Scientific,#A-11058),donkey anti-rabbit IgG 488 conjugate(Thermo Fisher Scientific,#A-21206)。
e.畸胎瘤形成实验
选取核型鉴定为正常染色体的iPSC扩增,EDTA消化,计数;用100µl PBS混悬诱导多潜能干细胞(细胞量约1×107),1∶1加入基质胶(每只小鼠注射1×107个细胞),植入NOD-SCID小鼠的一侧下肢腹股沟皮下,8~10周后处死小鼠,分离肿块将其置于甲醛溶液中固定,石蜡包埋切片,苏木精-伊红染色,光学显微镜下观察三胚层形成情况。
f.实时定量PCR(real time quantitative PCR,RT-qPCR)
首 先,收 集iPSC,加 入1 ml TRI试 剂(Sigma),混匀,-20℃过夜。利用Direct-zol RNA MiniPrep试剂盒(ZYMO RESEARCH)按照说明书操作抽提RNA。逆转录反应采用EasyScript™cDNA Synthesis Kit进行(20 µl体系),步骤同说明书。qPCR采用KAPA SYBR® Fast Mastermix实时荧光定量试剂盒依据说明书进行,反应体系如表1。

Table 1 qPCR reagents
qPCR反应程序如下。
阶 段1:95℃,3 min。阶 段2:95℃5 s,60℃30 s,40个循坏。阶段3:95℃10 s,65℃5 s。根据2-ΔΔCt方法,以GAPDH为内参,计算各个基因的相对表达量(PBMC作为阴性对照,H9-hESC作为阳性对照),引物序列见表2。

Table 2 Nucleotide sequences of primers used in RT-qPCR
g.支原体检测
收 取48 h培 养 液 上 清1 ml,2 000×g离 心10 min,去沉淀;17 000×g离心10 min,去上清,剩余底部50µl液体作为扩增样本模版;进行PCR,检测上游引物为ACACCATGGGAGCTGGTAAT,下游引物为CTTCATCGACTTTCAGACCCAAGGCAT及CTTCTTCGACTTCCAGACCCAAGGCAT。
h.整合分析实验
细胞DNA抽提具体步骤见前述,取P12的iPSC作检测;PCR实验具体步骤见前述;检测外源质粒载体整合,所用引物序列见表3。

Table 3 Nucleotide sequences of primers used in integrated analysis
2 结 果
2.1 临床特点
在婴幼儿期,2例患儿均以眼震为首发症状,主要表现为全面发育迟缓、肌张力低、步态异常。头颅核磁显示脑白质髓鞘形成低下。学龄前期,2例患儿肌张力正常,运动发育正常或轻度迟缓,语言发育均正常,Pt2头颅核磁显示40月龄头颅核磁内囊前肢髓鞘形成好转,其余区域脑白质仍表现为弥漫性低信号,提示髓鞘形成低下(图1)。Pt1进入学龄期后,步态恢复正常。临床特点对比见表4。

Fig.1 T2-weighted MRI images at 4 months(left)and 40 months(right)of Pt2

Table 4 Clinical characteristics of two patients with variants in TMEM163
2.2 遗传学特点
Pt1与Pt2分别发现TMEM163c.227T>G p.(L76R)与c.227T>C p.(L76P)新发错义变异,蛋白质结构预测显示(图2),在L76R突变体中,第76位的亮氨酸突变为精氨酸,改变了氨基酸携带的电荷量,突变体的氨基酸残基可能会破坏附近的泛素化位点,VENUS预测ΔΔG=2.3 kcal/mol,突变体蛋白质结构不稳定。在L76P变体中,第76位的亮氨酸突变为脯氨酸,从非芳香族变为芳香族氨基酸,更具极性,VENUS预测ΔΔG=2.7 kcal/mol,突变蛋白质结构不稳定,且可能破坏附近的泛素化位点。错译变异预测软件REVEL评估L76P为0.81,即有81%可能性为致病性变异,评估L76R为0.91,即有91%可能性为致病性变异。
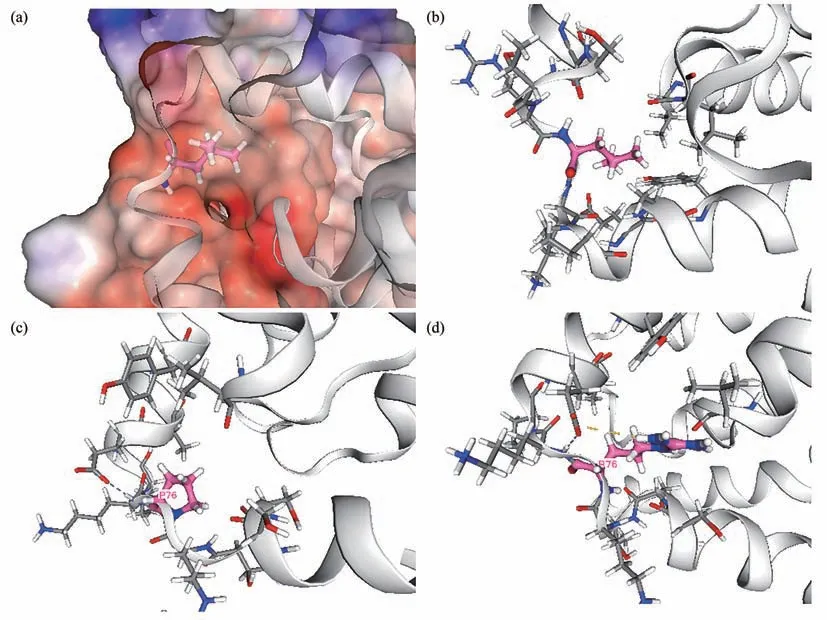
Fig.2 The predicted structure of TMEM163 wild-type and mutant proteins
Pt1检测发现TMEM163c.227T>G p.(L76R)新发错义变异,位点依据ACMG评级标准,评为“致病性”(PS1+PS2+PM1+PM2+PP3)。Pt2检测发现检测发现TMEM163c.227T>C p.(L76P)新发错义变异,位点依据ACMG评级标准,评为“致病性”(PS1+PS2+PM1+PM2+PP3)。
2.3 iPSC构建及鉴定
利用Pt2TMEM163c.227T>C p.(L76P)来源外周血单个核细胞构建iPSC。iPSC表现为典型的人胚胎干细胞形态学特征(图3a),碱性磷酸酶染色呈阳性(图3b)。经Sanger测序验证,其和患者血单个核细胞中突变位点一致(图3c)。核型分析结果无异常(图4)。免疫荧光染色证实其表达多能性标志物OCT4、SOX2、TRA-1-60和NANOG,且RT-qPCR分析证实其多能性标志物OCT4、SOX2、NANOG、LIN28和REX1在iPSCs中的表达高于PBMCs(图5)。此外,畸胎瘤实验证实其具有三胚层分化潜能(图6)。支原体特异性PCR检测结果表明,与阳性对照相比,诱导的iPSC没有支原体污染(图7a)。整合分析提示细胞没有外源质粒载体整合(图7b)。该细胞系已经过人类多能干细胞系全球注册中心(hPSCreg®,https://hpscreg.eu/)注册认证,细胞系编号为PUFHi00 4-A。

Fig.3 Morphology,alkaline phosphatase activity and Sanger sequencing of iPSC
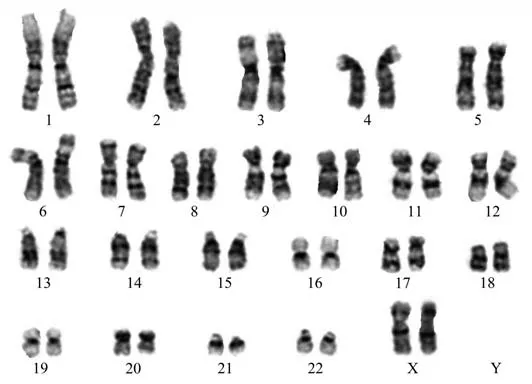
Fig.4 Karyotype analysis

Fig.5 Positive immunofluorescence staining for pluripotency markers

Fig.6 Representative hematoxylin and eosin staining of teratomas derived from the established iPSC clones

Fig.7 Mycoplasma test and integrated analysis
3 讨 论
HLD是因中枢神经系统髓鞘化低下导致的以发育迟缓为主要表现的一类遗传性疾病[11]。大多数重度髓鞘形成低下的患者在婴幼儿期起病,并发展为重度神经功能障碍,但部分患者症状较轻,症状在青春期或成年期开始出现。其主要特征为头颅MRI脑白质T2WI弥漫性高信号伴T1WI等信号、轻度低信号或轻度高信号。发病率预估范围为1/5 000~1/50 000[12],中国尚未见相关报道。临床表现为运动认知发育迟缓,神经系统异常如眼震、肌张力异常、共济失调、痉挛性截瘫及锥体外系征,部分患者可有牙齿发育异常等[13]。本研究中,2例患者临床特点为早期运动语言发育迟缓、头颅磁共振成像显示脑白质髓鞘化不良,且症状随生长发育逐渐改善。均于婴儿期起病,以眼球震颤为首发症状,学龄前期均有步态异常与肌张力低,临床诊断为HLD。与之前对2例患者的观察相比[2],Pt1于10岁时智力水平及运动发育正常,Pt2头颅核磁显示内囊前肢髓鞘化好转,均提示该类HLD具有独特的症状逐渐好转的特点。
目前有24种基因与HLD相关(OMIM,https://omim.org/),本研究病例为TMEM163同一位点2种错义变异,Pt1发现TMEM163c.227T>G p.(L76R)新发变异,Pt2发现TMEM163c.227 T>C p.(L76P)新发变异。两变异软件预测均提示有害,均未在千人数据库、ExAC数据库等收录,蛋白质结构预测提示突变体蛋白结构不稳定,且可能破坏附近的泛素化位点,引起细胞内蛋白质浓度失衡,导致少突胶质细胞功能障碍,影响髓鞘形成。功能实验证明两变异均可引起HeLa细胞内锌稳态失衡及斑马鱼髓鞘形成障碍[2],ACMG评级均为“致病性”(PS1+PS2+PM2+PP3)。据此,认为Pt1为TMEM163c.227 T>G p.(L76R)变异致病,Pt2为TMEM163c.227 T>C p.(L76P)变异致病,两例患者临床遗传学诊断为HLD。与先前评级相比[2],本研究首次对两位点进行蛋白质结构分析预测变异对蛋白质结构稳定性影响,首次纳入新软件预测结果(如PROVEAN、REVEL等),完善ACMG评级证据。
目前,HLD尚无治愈方法,其致残性为患儿家庭及社会带来了沉重负担,而探讨TMEM163独特临床病程的病理机制,尤其是髓鞘形成好转机制,有希望为同类疾病提供治疗线索。为此,本研究采集Pt2外周血构建iPSC。建立的患者iPSC在各方面与人胚胎干细胞相似,包括形态、增殖潜能、表面标志物、基因表达、染色体结构、变异位点、体外分化畸胎瘤形成、外源质粒整合分析等。经Sanger测序及染色体核型分析,其变异位点与患者血单个核细胞中变异位点一致且无大片段染色体结构变异,证明诱导过程未造成染色体结构损伤。干细胞标志物碱性磷酸酶染色阳性,经免疫荧光染色及RT-qPCR,与PBMCs相比,多能性标志物OCT4、SOX2、NANOG、LIN28和REX1表达升高,畸胎瘤分化实验可观察到肠、脂肪细胞、神经管等组织,证明其具有三胚层分化潜能。因采用质粒转染,故对外源质粒载体进行检测,结果提示细胞没有外源质粒载体整合。与大/小鼠模型相比,iPSC取材自患者,贴近人类遗传学背景,利于探索细胞通路、细胞代谢及转录因子相互作用机制。由iPSC诱导为脑类器官,可观察发育过程中的动态变化,为治疗探索奠定基础。iPSC诱导模型局限性为无法对行为学现象进行建模观察,在后续药物研究中,仍需结合动物模型观察行为学变化情况,以进一步明确药效。
4 结 论
本研究为国际首次对2例TMEM163致病HLD患儿进行随访研究,总结了该类HLD的自然病程,扩展了对HLD临床表型认识。国际首次构建了TMEM163c.227T>C p.(L76P)变 异 患 者 来 源iPSC,为机制研究及治疗探索打下了基础。
猜你喜欢
杂志排行

生物化学与生物物理进展的其它文章
- 42℃热疗抑制U251细胞增殖并诱导凋亡*
- The Differing Fortunes and miRNA Clusters Between Human Astrocytes and Neurons After Endoplasmic Reticulum Stress With Downregulation of EIF2B5*
- The Dynamic Changes and Clinical Significance of Serum Neuroglobin Levels in Patients With Acute Ischemic Stroke
- 组织蛋白酶B介导NLRP3小体在砷致小胶质细胞炎症激活中的作用*
- 一个白质消融性白质脑病家系新基因突变及临床表型研究
- 第五版《世界卫生组织中枢神经系统肿瘤分类指南》下成人胶质瘤诊断及预后*