胶质细胞病
——伴皮层下囊肿的巨脑性脑白质病致病机制研究进展*
2022-11-22王静敏
石 真 王静敏
(1)广州市妇女儿童医疗中心,广州 510000;2)北京大学第一医院,北京 100034)
伴皮层下囊肿的巨脑性白质脑病(megalencephalic leukoencephalopathy with subcortical cysts,MLC)是一种罕见的中枢神经系统髓鞘变性病,是由于MLC1或胶质细胞黏附分子(glial cell adhesion molecule,GlialCAM)基因突变所导致的常染色体显性或常染色体隐性遗传病,其病理特征是星形胶质细胞肿胀及髓鞘外层囊泡形成。MLC的典型临床表现是出生后第1年内出现巨颅畸形、中度至重度运动发育迟缓、智力正常或轻度智力发育迟缓,随时间进展运动功能逐渐恶化,临床出现共济失调、癫痫发作、痉挛性截瘫和锥体外系异常的表现[1-2]。患儿颅脑磁共振成像(magnetic resonance imaging,MRI)显示双侧大脑白质对称弥漫性异常高信号,伴或不伴前颞叶和额顶叶皮质下囊肿[2]。MLC1(OMIM 605908)是由Van der Knaap等于2001年发现的MLC的第1个致病基因[3],MLC1突变患儿临床表现为经典型MLC,即MLC1型(OMIM 604004),占MLC患者的75%。GlialCAM于2011年被确定为MLC的第2个致病基因[4],与GlialCAM突变相关的疾病表型被命名为MLC2型。MLC2型按照遗传方式的不同,可进一步分为2种,常染色体隐性遗传的MLC2A型和常染色体显性遗传的MLC2B型,MLC2型的患者占MLC患者总数的20%。MLC2A和MLC2B呈现不同的临床症状和预后,MLC2A型与MLC1型运动功能随年龄增长逐渐恶化,而MLC2B型又称为改善型,部分患者1岁以后出现临床及头颅MRI的改善,甚至完全恢复正常[4]。到目前为止,国际报道了约400例MLC患者,其中89例为MLC2型患者[5-7]。其中,北京大学第一医院团队报道了国内首例MLC1及GlialCAM突变导致的MLC患者[8-9],是目前国内报道MLC患者最 多 的 单 中 心[5-6],包 括MLC1型 患 儿24例、MLC2A型患儿1例、MLC2B型患儿5例。患儿头颅MRI表现、脑组织病理切片及既往体内和体外实验表明,MLC发生的致病机制与水和离子稳态失衡密切相关[10-11]。
1 MLC与GlialCAM的表达及结构
MLC1所编码的MLC1蛋白是一个由377个氨基酸组成的8次跨膜结构的蛋白质[3]。MLC1在人脑组织中在位于血管周围的星形胶质细胞的远端突起、室管膜下和软脑膜下区域以及小脑的伯格曼胶质细胞中表达[12]。MLC1作为跨膜蛋白,与细胞膜或基质中蛋白质有直接或间接相互作用,例如Na+/K+-ATP酶的β亚基、内向整流钾通道4.1(inward rectifier-type potassium 4.1,Kir 4.1)通道、水通道蛋白4(aquaporin protein-4,AQP4)、肌营养不良蛋白、小窝蛋白1和瞬时受体电位香草精4阳离子通道(transient receptor potential vanilloid-4 cation,TRPV4)[13]。MLC1突变后影响其他蛋白质的活性,例如体积调节的阴离子通道(volume regulated anion channel,VRAC)[11]、LRRC8[14],影响肿胀后星形胶质细胞体积的恢复参与MLC的发生。GlialCAM编码的GlialCAM蛋白是由416个氨基酸组成的跨膜蛋白,具有典型的免疫球蛋白样细胞黏附分子结构,分为胞外区、跨膜区和富含脯氨酸的胞内区,胞外区含有1个信号肽和2个免疫球蛋白样功能域[4]。CAM通过复杂的细胞信号转导系统对细胞的迁移、增殖、分化与凋亡存活起着重要作用。GlialCAM胞外区与临近细胞胞膜上的黏附分子及胞外基质相互作用,在胞内与细胞肌动蛋白细胞骨架相互作用,肌动蛋白细胞骨架在维持细胞的完整性和形态上十分重要。在中枢神经系统,GlialCAM除了与MLC1共定位于星形胶质细胞终足处外,还存在血脑屏障处、小脑伯格曼细胞及轴索中及髓鞘中。在髓鞘中,GlialCAM位于轴索与髓鞘的接触面,以及形成髓鞘中,并与神经丝蛋白重链共定位[4]。GlialCAM作为MLC1的分子伴侣,协助MLC1在细胞膜的正确定位,并将其集中 在 细 胞-细 胞 连 接 处[4,15]。目 前 已 发 现 的GlialCAM的大多数错义突变位于第1个Ig结构域(IgV型),并影响GlialCAM向细胞-细胞连接处的定位,同时影响突变的GlialCAM与正常GlialCAM蛋白之间的相互作用,而位于第2个Ig结构域(IgC2型)的其余突变不影响GlialCAM在细胞-细胞连接处的定位。通过模型分析GlialCAM可通过第86~92个氨基酸残基形成特定互相作用片段,从而在相邻细胞间形成反式相互作用[16]。在IgV区的表现为,显性遗传突变的GlialCAM可以挽救不同位点显性遗传突变的GlialCAM,使其在细胞-细胞连接处的表达量增加,但是不能挽救隐性遗传突变的GlialCAM在细胞内的转运,野生型GlialCAM不能起到挽救显性遗传突变GlialCAM细胞内转运的作用,从生物物理学层面解释了GlialCAM突变会导致常染色体隐性遗传(AR)和常染色体显性遗传(AD)两种不同遗传方式的基础。
2 水离子稳态与平衡
早期在对MLC1型患者尸检脑组织病理学检查显示脑白质中有大量空泡存在,电镜下观察发现髓鞘崩解及髓鞘内空泡形成[12,17-18],细胞学实验观察到的星形胶质细胞空泡/肿胀以及在MLC疾病模型中报道的K+、Cl-等电流的变化、星形胶质细胞自身调节能力障碍,导致水和离子稳态失衡,引发MLC。调 节 性 容 积 减 少(regulated volume decrease,RVD)是动物细胞调节自身体积的基本能力,在低渗刺激时,星形胶质细胞可以通过RVD机制,在瞬时渗透性肿胀后,通过K+、Cl-外流来实现重新调节其体积,以维持细胞形态和正常功能[19]。ClC-2是一种广泛表达的细胞膜的氯离子通道,在超极化、细胞肿胀时缓慢激活[20]。ClC-2、GlialCAM及MLC1在星形胶质细胞终足处共定位,形成三聚体结构[21],为ClC-2参与MLC的发生提供了空间上的可能。研究发现,星形胶质细胞MLC1敲除会导致GlialCAM和ClC-2在少突胶质细胞中错误定位,而GlialCAM纯合敲除或点突变后影响MLC1及ClC-2在细胞-细胞连接处的定位[22],并影响ClC-2通道的电容传导性。ClC-2的N端通过与GlialCAM细胞内特定环状结构相互作用从而转运至细胞膜处[22]。在细胞内异源共表达后,GlialCAM增加ClC-2电流,而GlialCAM纯合敲除及点突变小鼠中,ClC-2内向整流电流减少[23],但ClC-2敲除后并不影响GlialCAM和MLC1在细胞膜处的定位[22]。进一步研究发现,在GlialCAM纯合敲除的小鼠模型中,通过与选择性开放ClC-2通道的转基因小鼠杂交并不能挽救空泡表型[24];在GlialCAM和ClC-2双重敲除的小鼠脑组织,比仅敲除ClC-2的小鼠形成更严重的髓鞘空泡表型,均表明ClC-2功能障碍仅作为致病因素的一部分参与MLC的发生[22],GlialCAM突变后影响其他过程共同参与致病仍需进一步研究。
3 细胞间通讯
MLC的最常见的致病基因为MLC1,在星形胶质细胞中特异性表达,而异常的病理表型主要见于少突胶质细胞形成的髓鞘[10,12],既往研究发现星形细胞功能异常也可导致髓鞘结构异常、巨颅及头颅影像学的异常表现,如胶质纤维酸性蛋白(glial fibrillary acidic portein,GFAP)基因突变导致亚历山大病[25]。此外,星形胶质细胞通过分泌细胞因子和生长因子促进髓鞘的形成[26],通过缝隙连接与少突胶质细胞形成异源性相互作用[27]。结合患儿弥漫性脑水肿为特征的头颅MRI表现、脑组织的切片及细胞学实验观察到的星形胶质细胞空泡/肿胀以及在MLC疾病模型中报道的K+、Cl-和Ca2+电流的变化(图1)[11,20,22,28],提示MLC1突变可能直接或间接影响细胞间通讯和胶质细胞合胞特性,最终导致MLC患者少突胶质细胞的病理改变。

Fig.1 Putative model of the mechanism of formation of the ternary complex between GlialCAM,MLC1 and ClC-2,mutated GlialCAM leads to the reduced conductivity of ClC-2图1 GlialCAM突变后通过MLC1/GlialCAM/ClC-2三聚体影响ClC-2通道效率导致星形胶质细胞肿胀的可能机制示意图
胶质细胞的合胞体指胶质细胞通过一系列细胞间连接蛋白形成广泛耦合,允许水、离子和大分子进行快速空间再分配,使神经系统在突触活动后恢复稳态状态,这些分子蛋白包括缝隙连接蛋白、水和离子通道、膜转运蛋白以及与其相关的结构蛋白复合物[29]。细胞连接蛋白是一个由20多个蛋白质组成的家族,构成了细胞缝隙连接的通道[30]。这些通道直接连接相邻细胞的细胞质,可允许离子和一部分小分子物质渗透通过。在神经元中,它们参与构成电突触,而在胶质细胞中,它们提供信号转导功能[30]以及维持内环境平衡[31]。有髓神经胶质细胞上普遍表达多种连接蛋白,并且连接蛋白是中枢神经系统和外周正常髓鞘形成所必需的。其中,缝隙连接蛋白32(connexin32,Cx32)基因的部分突变可导致中枢神经系统脱髓鞘的疾病[32]。动物实验表明,仅敲除少突胶质细胞缝隙连接蛋白47(connexin47,Cx47)基因或Cx32基因的大鼠可以存活和繁殖,但是同时敲除Cx47或Cx32的大鼠会出现严重的中枢神经系统脱髓鞘,并伴有震颤和强直性痉挛发作,提示少突胶质细胞间通讯对维持髓鞘完整性及功能有重要作用。Lutz等[33]研究了星形胶质细胞特异性敲除Cx43和缝隙连接蛋白30(connexin30,Cx30)基因对少突胶质细胞成熟和髓鞘生成的影响,发现双敲除小鼠显示出水肿的星形胶质细胞、空泡状少突胶质细胞和髓鞘肿胀,这种表型在出生后第23天开始出现并持续到成年。此外,星形胶质细胞中缺乏Cx43和Cx30与髓鞘形成受损有关,如导致髓鞘碱性蛋白(myelin basic protein,MBP)表达减少。Cx32与Cx47在少突胶质细胞及星形胶质细胞间有共定位。因此,提出星形胶质细胞特异性间隙连接的缺失会导致少突胶质细胞和髓鞘病变[33]。Magnotti等[34]研究了星形胶质细胞连接蛋白对少突胶质细胞的影响,发现Cx43-/-/Cx32-/-小鼠呈现出髓鞘空泡形成的表型。与野生型相比,在这些小鼠中观察到星形胶质细胞数目显著减少。这些研究提示了星形胶质细胞之间、星形胶质细胞与少突胶质细胞之间连接异常,胶质细胞合胞功能异常是导致水肿形成和髓鞘损伤的原因。既往研究发现,Cx43定位到星形胶质细胞细胞膜需要GlialCAM的参与[35]。近期研究表明,GlialCAM诱导Cx43在细胞膜上的定位和稳定后,MLC1通过抑制/降低ERK1/2激酶的Cx43磷酸化,从而促进Cx43在细胞膜上的稳定性,阻碍Cx43的内化和通道关闭,从而维持星形胶质细胞间的缝隙连接通讯[36],突变的MLC1通过ERK1/2的激活促进Cx43磷酸化,促进Cx43的内化,减少在细胞膜处形成缝隙连接(图2),同时细胞间通讯效率下降,表明星形胶质细胞中Cx43受到MLC1调控以及Cx43参与MLC的发病机制,提示了潜在的治疗干预的靶向途径。
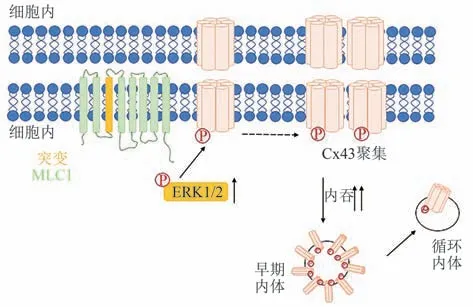
Fig.2 Putative model of the mechanism of mutated MLC1-mediated Cx43 recycling in astrocyte图2星形胶质细胞中MLC1突变介导的Cx43循环调节的可能机制示意图
4 结语与展望
MLC是婴儿期起病的中枢神经系统的髓鞘变性病,其致病基因为MLC1或GlialCAM。根据临床表现分为MLC1及GlialCAMAR遗传方式致病的经典型(MLC1型及MLC2A型),该类分型运动功能逐渐恶化,最终至痉挛性瘫痪,而GlialCAM突变引起的AD遗传方式致病的患儿为改善型(MLC2B型),部分患儿1岁以后出现临床及头颅影像学改善的情况,但有部分以AD遗传方式致病的MLC患儿表现出孤独症样改变,该类患儿临床无改善出现的机制仍待进一步研究;AD突变的GlialCAM在细胞膜的定位可以被不同AD突变的GlialCAM所挽救,但不能被野生型蛋白所挽救[16],是显性副性效应的体现,而既往推测改善型患者在1岁之后出现临床好转的原因可能是与MLC1与GlialCAM在发育过程中时空特异性的表达相关,最近在生物物理学层面针对突变及蛋白质结构的分析和预测,为发病机制的研究及明确GlialCAM不同突变位点遗传方式的分类[16]提供了新的分析思路。患儿头颅MRI表现出弥漫性脑水肿的特征,细胞学实验观察到的星形胶质细胞空泡/肿胀、髓鞘崩解及囊泡形成,提示星形胶质细胞功能障碍,水和离子转运失衡,引起少突胶质细胞功能障碍,导致MLC的发生;星形胶质细胞之间、星形胶质细胞与少突胶质细胞之间通过缝隙连接形成合胞体,在调节神经系统内环境稳态中发挥重要作用,提示MLC1/GlialCAM突变可能通过直接或间接作用,影响细胞间通讯和胶质细胞合胞功能,最终导致MLC特征性病理改变的发生。MLC作为脑白质病中患病率相对较高的罕见病,其致病机制的深入研究为其他以白质肿胀为特征的脑白质病,提供致病机制研究的可能思路,同时为将来的精准治疗提供研究基础。
综上,MLC1/GlialCAM突变后,通过影响ClC-2通道、细胞连接蛋白Cx43,影响胶质细胞合胞功能,导致水和离子稳态失衡是MLC病理中星形胶质细胞肿胀及髓鞘囊泡形成的重要原因,其具体机制仍有待实验进一步研究证实。
