少突胶质细胞病
——佩梅病的临床特点及致病机制*
2022-11-22段若愚延会芳王静敏
段若愚 延会芳 王静敏**
(1)北京大学第一医院儿科,北京 100034;2)国家医学儿童中心,首都医科大学,北京儿童医院神经内科,北京 100045)
收稿日期:2022-08-10,接受日期:2022-09-28佩梅病(Pelizaeus-Merzbacher disease,PMD)是一种以髓鞘形成低下为特征的X连锁隐性遗传的脑白质病。1885年,德国医生Friedrich Pelizaeus最先描述了一个表现眼震、发育落后、痉挛性截瘫及共济失调为主要表现的家系[1];1910年,Ludwig Merzbacher[2]发现此病呈X连锁隐性遗传的方式,佩梅病的名字由此得来。此外,Ludwig Merzbacher指出,佩梅病患者脑活检显示白质髓鞘严重脱失,其脑白质髓鞘化不良的典型特征逐渐明确。据此,佩梅病又叫做髓鞘形成低下性脑白质营养 不 良I型(hypomyelinating leukodystrophy disease 1,HLD1)。
1 PMD流行病学及病理学特点
1.1 PMD流行病学特点
PMD多在婴幼儿期起病,男性多于女性,其发病率在白种人群(德国、英国及美国犹他州)中为0.97/100 000活产婴儿,在东亚人群中为1.45/100 000男性活产婴儿[3-4],目前在国内尚缺乏相关研究。
1.2 PMD病理学特点
PMD患者脑部病理学特点为整个中枢神经髓鞘发育不良及白质萎缩伴星型胶质细胞的增生、皮质下轴突丢失、残存的轴突呈球状[5]。在脊髓中,丘脑脊髓束和前脊髓小脑束相对较少,而皮质脊髓束、背侧束、背外侧束和背侧脊髓小脑束明显有髓鞘异常[6]。脑萎缩主要为胼胝体萎缩,部分可累及端脑、小脑等。其次,PMD患者脑组织中出现神经元细胞的缺失,包括海马、小脑、黑质-纹状体及丘脑神经元的缺失。其中以小脑神经元缺失最为常见。“髓鞘岛”在不同患者中形状不同。相应地,严重表型的患者脑白质髓鞘可完全缺失,反之相对轻型患者中央白质部白质髓鞘呈典型的“虎斑样”外观[5]。其组织细胞学染色显示:PMD患者脑部星型胶质细胞的细胞质中发现弥散分布的非常小的中性脂肪滴,其发病机制可能涉及髓鞘脂蛋白或蛋白脂类。偶有血管周围间隙含有少量巨噬细胞,且巨噬细胞内充满大量嗜苏丹红细胞小滴,但PMD是否能被归类为嗜苏丹型脑白质营养不良仍存在争议。
2 PMD临床特点
2.1 PMD临床表现
PMD作为最具代表性的HLD,其主要特征为头颅磁共振成像(MRI)显示脑白质髓鞘化不良,即T2WI脑白质弥漫性高信号[7]。临床表现包括婴儿早期或幼儿期出现的眼震、肌张力低下、智力运动发育落后伴或不伴共济失调、癫痫发作及痉挛性截瘫等[8]。各种临床表现在不同的患者中呈现出不同的表现方式,甚至同一个家系的患者也会呈现出不同的临床症状。患者病程长短不一,部分患者婴儿早期即死亡,部分患者病程长,病情进展缓慢甚至最终寿命长短并不受影响。眼震通常在出生后1年内出现,随着神经系统的成熟,部分患儿可同时出现喉喘鸣及癫痫发作[9],精神运动发育迟缓在2岁内出现且运动发育较智力发育迟缓更突出。肌张力障碍及共济失调通常在患者6个月到2岁之间出现,1岁之后出现腱反射亢进,2~4岁间出现痉挛,其中共济失调可持续到8岁[10]。视神经萎缩是一种常见但具有异质性的表型,该症状通常在6岁左右被发现,视诱发电位低振幅,波形异常伴潜伏期延长[11],部分患者同时存在听觉脑干诱发电位异常(低波幅等)[12],前庭功能障碍及眼震电图异常。最初的研究认为,相对于其他脑白质异常,PMD患者不伴有周围神经的异常,但腱反射减弱或消失、周围神经传导速度异常、明显的肌萎缩及四肢冰冷等症状在Grossi等[13]的研究中被证实。
2.2 PMD诊断标准及分型
PMD的临床诊断标准早在1885年和1910年在Pelizaeus及Merzbacher报道的家系中详细阐述。婴儿期或儿童早期起病,临床表现包括眼震、锥体束征及肌张力障碍等。中央白质呈髓鞘障碍的斑片状分布,髓鞘岛保存完好,形成典型的“虎斑”外观,轴突相对保存完好。此表型随后被划归为PMD I型-经典型。1954年报道了一类临床表现更为严重的患者,这些患者发病早,从来没有神经系统各项功能的获得,大脑中髓鞘完全缺失,中央髓鞘可能完全缺失,少突胶质细胞数量减少,部分出现形态学改变,偶尔可见少量、薄而不规则的髓鞘“微岛”[14]。电镜下,髓磷脂压实和层状排列缺陷,该表型被归为PMD II型-先天型或Seitelberger型。与此同时,根据PMD临床病理学的特征,研究者发现了一群临床表现差异较大的患者,他们成年期发病,不存在精神运动发育迟缓、身材矮小、不自主运动等PMD综合征的特点,但有明显的全身肌肉萎缩及去神经支配征象,感觉障碍及胸腰椎异常及尾骨发育不全。神经病理学特征同时存在“虎纹样”脱髓鞘模糊斑片状外观,弥漫性轴突缺失。但不同的是,该类患者同时存在腹角细胞缺失和周围神经脱髓鞘以及锥体束、小脑和视束的退行性变,并伴有视束退行性变。此型被归为成人型/Liiwenberg-Hill型[15],由于成人型不具有PMD综合征典型的症状,该表型后被称为痉挛性截瘫II型(spastic paraplegia-2,SPG2)。根据PLP1相关综合征神经病理学特征、起病及病程特点,Seitelberger将PLP1谱系障碍综合征分为先天型、中间型、经典型及无PLP1综合征4种(表1)[6]。
但事实上,研究者发现,在缺乏相关的酶或其他生物标记物的情况下,此时期的临床分型很大程度同时依赖神经病理学结果[2,14],临床上很多表型具有同质性的个体,神经病理学结果却是非特异性的[12],如髓鞘化的程度或分布等。Boulloche等[11]指出明确的遗传方式和一致的临床特征的关联能更好地定义髓鞘功能障碍性疾病,且随着头颅核磁影像学更好地应用于临床并逐渐代替神经病理学反映脑髓鞘化状态,PMD的诊断标准更新为:a.躯干肌张力减退;b.生后1个月左右出现眼震,眼震随着年龄增长逐渐好转或消失;c.1岁以内出现椎体征、肌张力障碍及小脑征的进展;d.智力运动障碍缓慢进展,且运动相较于智力障碍更严重;e.神经电生理及磁共振成像结果中髓鞘异常仅局限于中枢神经系统[11]。鉴于共同的遗传学背景,但临床表现不同,SPG2被划归为与PMD同一疾病谱系的等位基因病,即为PLP1谱系障碍综合征[16]。但随着越来越多的PMD患者被发现,其临床表现多样性使研究者认识到以往分类方法并不能有效体现PMD患者之间的异同。Cailloux等[10]提出,根据PMD患者能获得的最高运动水平,将PLP1谱系障碍综合征分为具有连续统一性的5型(Forms 0-4):0类患者表现最重,没有任何运动能力的获得;1型次之,仅能获得抬头的能力;2型能独坐;3型能扶站;4型患者对应临床表现最轻的SPG2型,能够独立行走[10,17]。但此分类在临床和研究中的实用性需要进一步评估。
3 PMD的遗传学特点
3.1 致病基因PLP1及蛋白脂蛋白I型
PMD的致病基因为位于Xq22.2的蛋白脂蛋白(proteolipid protein,PLP1,NM_001128834.20)基因[18],全长~15 kb,包含7个外显子,编码277个氨基酸。PLP1编码两种主要的髓鞘脂蛋白,蛋白脂蛋白(proteolipid protein,PLP)I型及其剪接体DM20。PLP1是一种4次跨膜蛋白(图1),包括2个胞外环(extracellular domain,ED)、4个跨膜区(transmembrane domain,TMD)、3个胞内环(cytoplasm domain,CD),第二胞内区116~150位氨基酸形成PLP区(PLP-speciffic region),存在于PLP1蛋白中,不包含该区域的剪接体即为DM20[19]。跨膜区由4个跨越双层膜结构的α螺旋(TMD1-4)组成,胞外环及胞内环连接各个螺旋形成PLP拓扑结构(图1)。
PLP1在内质网(endoplasmic reticulum,ER)中合成,运输到细胞表面与脂质形成髓鞘膜。同时是“DM”分子家族中的一员,该家族成员具有高度的序列保守性,特别在跨膜区域及半胱氨酸基序的位置[20]。PLP1中含有14个半胱氨酸残基,4个半胱氨酸通过二硫键连接,Cys200-Cys219和Cys183-Cys227,位于细胞第二胞外区,以形成含有二硫键的环状结构。位于5、6、9、108、138和140位的6个半胱氨酸残基与长链脂肪酸(主要是棕榈酸)交联形成硫酯锚定在细胞膜上(图1),其余4个定位在24、32、34和168位为游离巯基[21]。研究表明,野生型PLP1(WT-PLP1)最初可以单体或二聚体的形式存在于内质网中,经过蛋白质亚基的交联组装,在成熟期形成稳定的寡聚体表达在细胞表面的髓鞘中[22-23]。PLP1寡聚体在内质网中的折叠加工及运输到质膜需要一定的时间,过快的寡聚化可影响其撤离出内质网从而影响其在细胞内的运输[23]。
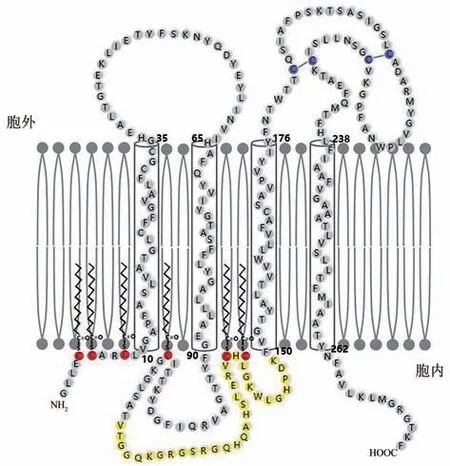
Fig.1 Schematic diagram of quadric transmembrane structure图1 PLP1 4次跨膜结构模式图
PLP1和DM20是中枢神经髓鞘形成的主要蛋白质(约占50%),目前认为PLP1主要在髓鞘形成中发挥作用,包括髓鞘的膜黏附和致密髓鞘内线的形成[24]。在发育早期少突胶质细胞前体细胞阶段,DM20表达占优势[25]。PLP1参与少突胶质细胞的成熟,少突胶质细胞-轴突相互作用的早期阶段和轴突的包裹,轴突的维持和存活等。Kitagawa等[26]认为PLP1也能发挥通道蛋白的作用。此外,PLP1的表达与分泌一种增加少突胶质细胞数量的因子直接相关[27]。
3.2 PLP1突变类型
引起PMD的PLP1突变类型包括重复突变、点突变、小片段插入及缺失突变[28],其中PLP1重复突变人数最多(占60%~70%)[29-30],点突变次之(占25%~30%),小片段插入及缺失最少(<5%)[31]。PLP1重复突变涉及片段大小不等,通常为PLP1基因与临近基因构成的大片段串联在一起。此外,部分重复片段可易位到X染色体的其他区域或发生片段内的断裂、倒位等[32]。PLP1点突变中主要为错义突变,无义突变、移码突变及剪接位点等无功能突变则主要对应较轻的SPG2表型[16]。
4 PMD的基因型表型关系
PLP1基因座上所有突变类型均可导致一定的疾病表型,不同的突变类型在患者中表现出不同的表型。PLP1重复突变涉及基因的数量和种类多样,以往研究中该变异类型与PMD表型的关系说法不一。Regis等[33]研究认为,PLP1重复突变片段的长度与患者的表型的轻重没有关系,同时发现每个重复片段中在低拷贝重复序列(low copy repeats,LCR)中或周围都有两个断点,但断点对PMD临床表现的影响并无进一步的阐述。而随后对50名PLP1重复突变的PMD患者的研究则认为,PLP1重复片段的长度与PMD患者表型的严重程度具有密切关系,可能反映重复片段中的其他剂量敏感基因在疾病中发挥了一定作用[32],包括与神经发育有关的CASK和IL1RAPL2[34-35],含有这两个基因的PLP1重复片段会产生更严重的PMD表型[36-37]。同样的,虽然PMD为X连锁遗传的遗传方式,PLP1重复突变携带者的女性患者在临床上时有报道,其表型的严重与否与重复片段中剂量敏感基因的数目也密切相关[32]。
PLP1点突变相关的PMD患者表型囊括了最严重的先天型到最轻的SPG2型。研究者最初通过构建患者变异来源的转基因动物模型,揭示不同PLP1点突变产生轻重不同水平的表型:Plpjp(jimpy)小鼠模型是表型最严重的动物模型之一,该突变体的特征是242位丙氨酸取代了缬氨酸,小鼠在疾病早期表现出严重的髓鞘化不良、震颤等并在30 d左右死亡,其表现符合PMD临床上最严重的先天型[38]。而相应的Plpjp-rsh(“rumpshaker”)小鼠的表型较为温和,为186位异亮氨酸发生了苏氨酸的突变,其特点是仅限于后腿的震颤,震颤在30 d左右达峰值且持续终生。Plpjp-rsh不伴有癫痫,其髓鞘形成低下相关的临床特征不明显,生存期不受影响甚至可存活下来成功繁殖。该类小鼠的表现类似于临床上最轻的SPG2型,其成熟的少突胶质细胞数量增加,即使这些小鼠是整体呈现低髓鞘化的[39]。髓鞘形成开始是延迟的,随着动物年龄的增长,先前裸露的轴突获得了一些髓鞘。髓鞘成分有轻微的异常,免疫印迹显示PLP1减少,而DM20蛋 白 水 平 保 持 不 变[40]。而md(myelin defixient)大鼠模型,为74位苏氨酸突变为脯氨酸的突变体,大鼠生后出现严重的肢体震颤,并伴有癫痫,所有症状在10 d左右开始明显,并在3~6周间死亡。相应地,Plpjp-msd小鼠脑内虽然少树突细胞数量增加但多为未成熟细胞,未成熟少突胶质细胞凋亡率增加,成熟少树突细胞数量减少[38]。合成的异常蛋白质对少突胶质细胞存在毒性作用,到达髓鞘内的PLP1蛋白存在功能异常。相对于点突变,PLP1小片段的缺失或插入通常产生相对较轻的表型,但最近的一项研究在PLP1的3号内含子(NM_000533.5:c.453+59_+259del)中发现了一个新发的深部内含子缺失,患者的临床和影像学符合较重的先天型,这可能是由于该突变类型导致PLP1异 常 剪 接[41],Cloake等[42]研 究 提 出Leu30Val突变可以在该患者中产生几种新的免疫原性表位,最终可能导致一小部分患者发展为多发性硬化(multiple sclerosis,MS)。
对PLP1结构的研究表明,相对于发生在蛋白质表面的变异,跨膜区域的变异更能被容忍,ED2环的缩短和相应CD2环的延长也可能会阻止正确的折叠和相互作用[43],从而产生较严重的临床表现。而PLP1特异性区域的变化通常导致较温和的表型。此外,编码PLP1和DM20亚型的进化上保守残基的变化被认为是导致严重表型的原因[10]。
对已报道变异类型及临床信息进行关联分析,结果表明PLP1点突变可产生PLP1疾病谱系的所有表型(Form 0-4),PLP1重复突变主要产生较温和的表型(Form 2,3),而无义、移码突变及小片段的缺失等导致PLP1编码的氨基酸提前终止的一系列突变,则通常会表现为最轻的表型(Form 3,4)(图2)[8]。

Fig.2 The relationship between genotype and phenotype of PLP1 disease spectrum syndrome图2 PLP1疾病谱系综合征基因型和表型关系示意图
PMD是一种X连锁隐性遗传的疾病,患者家族中携带者女性通常是也本应该是无症状的。然而,由于X染色体随机失活,当携带突变的那条染色体发挥主导作用时女性携带者便可发病。该类患者主要表现为神经系统的症状,一般起病较晚,但表型比家族中的男性患者轻,因轻度的痉挛性截瘫起病,到晚年逐渐发展为进行性脑白质营养不良伴痴呆[44-45]。
5 PLP1突变致病的细胞学机制研究
5.1 PLP1与髓鞘
中枢神经系统的髓鞘由少突胶质细胞形成,在大脑髓鞘发育中,大脑室下细胞产生一定量少突胶质细胞前体细胞(oligodendrocyte progenitpr cells,OPCs),OPCs在大脑中分裂、分化、迁移,最终形成髓鞘前少突胶质细胞(pre-myelinatinating oligodendrocytes,Pre-OLs)。Pre-OLs进一步成熟,其细胞膜以紧密的螺旋状层层包裹在中枢神经系统(central nervous system,CNS)轴突细胞膜上,形成髓鞘[46]。髓鞘由蛋白质、脂质与水分构成,成熟髓鞘中15%~20%为蛋白质,而PLP1和DM20约占髓鞘蛋白的50%[17]。髓鞘碱性蛋白(myelin basic protein,MBP)结合到膜的细胞质表面,将细胞质表面紧密相连,使髓鞘在轴突周围形成紧密的螺旋。而PLP1作为4次跨膜膜的疏水蛋白,它在跨膜α螺旋3和4之间包含一个相对较大的细胞外环,这被认为是介导髓鞘膜外表面之间的相互作用,再次导致膜的紧密堆积(图3)[47]。髓鞘的其余成分中60%~75%为脂质,其中胆固醇约占25%,而PLP1与胆固醇结合最终在膜上组装成髓鞘。髓鞘具有绝缘作用,其上的朗飞氏结可使神经冲动跳跃传递,提高神经冲动的传导速度,并具有轴突保护作用[48]。由此,髓鞘在神经信息精确、高效传递及中枢信息整合中发挥重要的作用。

Fig.3 Longitudinal section(a)and transverse section(b)of myelin sheath structure图3髓鞘结构纵切面(a)及横切面,(b)示意图
髓鞘正常结构的形成及功能的维持,需要PLP1通过适当的途径不断转运到质膜上与胆固醇装配形成。其转运可以通过两种途径:一种通过“脂质筏(lipid rafts,LF)”的分选,此种方式为PLP1的主要上膜方式。Simons等[49]研究表明,髓鞘膜中的胆固醇和糖脂在内质网和反式高尔基体网络中与其他脂质分离,在膜内形成小的结构域,就像漂浮在其他脂质海洋中的筏,叫做“脂质筏”。PLP1在内质网翻译加工,接下来转运到高尔基体进行分选,通过囊泡分泌到达细胞膜,随后回到晚期内体/溶酶体中。晚期内体/溶酶体是PLP1的“动态调节池”,可以将其储存的PLP1通过囊泡转运到达细胞膜,最终形成髓鞘[46,50]。另一种途径为PLP1通过自身流动直接到达细胞膜表面,与胆固醇及其他膜蛋白形成髓鞘,此种途径为次要运输途径(图4)。

Fig.4 Mechanism of PLP1 mutation affecting PLP1 to the plasma membrane图4 PLP1突变影响PLP1上膜的作用机制示意图
总之,由于成熟的髓鞘为致密的含水量较少的结构,而髓鞘发生到成熟中的任何一个环节出现问题都可能导致髓鞘发育不良,表现为髓鞘含水量增高,脑核磁T2WI上脑白质呈弥漫性高信号影。而PLP1突变就是通过影响髓磷脂结构从而影响髓鞘的成熟[51]。
5.2 PLP1突变影响髓鞘结构及功能的作用机制
5.2.1PLP1点突变的作用机制
PMD相关的PLP1点突变主要为错义突变,此外部分点突变导致临床症状较轻的SPG2。错义突变可以发生在PLP1的所有编码区,每种突变不仅在不同家系中有不同的临床表现,其产生的致病机制也不尽相同。
位于PLP1及DM20保守区域的突变可导致较严重的表型,而发生在PLP1特殊区域的点突变主要导致SPG2,这可能与在后者产生的突变体仍保有完整的DM20蛋白有关[16]。同时缺乏这两种同工型的Plp基因敲除小鼠的髓鞘内线(intraperiod line,IPL)有缺陷[24],后期可伴有轴突病。仅表达一种同工型的基因敲除小鼠仍保持异常,而当同时存在两种同工型时,可防止异常表型[52]。由此,PLP1/DM20两种同工型的正常表达对于维持髓鞘结构与功能都是必须的[53]。
以往研究表明,PLP1/DM20以寡聚体的形式存在于成熟髓鞘中,寡聚化也是膜蛋白退出内质网的必要条件[54]。虽然WT-PLP1在较长的成熟期后在细胞表面形成稳定的寡聚物,但突变形式的PLP1可通过异常寡聚化而致病。a.一些致病点突变导致异常PLP1寡聚体形成可能是由于发生了影响单体整合的突变,使得通常被正确折叠的蛋白质掩盖的“黏性”螺旋面变得表面可接近,从而导致蛋白质寡聚体的异常组装[55]。PLP1的寡聚化涉及TMD序列中特异性的螺旋-螺旋相互作用基序[56]。例如,PLP1 TMD4螺旋存在各种致病突变的位点(A241P、A242V、A242E、G245A、G245E、A246T、A247E、A248P和S252F[57]),其中突变程度很小的残基(G、A和S)位置无处不在,这种小的侧链残基在螺旋-螺旋二聚和包装中发挥重要作用:通过小残基之间相互作用基序的介导,螺旋与螺旋间的自我结合形成。在完整的PLP1单体膜结构域中,促进其折叠的螺旋间相互作用可能消耗其中几个基序,因此一旦PLP1单体正确折叠,很可能只有保留在结构域表面的基序可用于介导蛋白质寡聚。而PLP1突变体可通过暴露正常折叠蛋白“隐藏”的基序从而引发蛋白质的异常组装。b.在内质网处过快组装成寡聚物而致病[23]。Swanton等[23]研究表明,与WT-PLP1相比,PLP1点突变体在在内质网中更快地形成寡聚体。TMD4(或其他TM α螺旋)面的暴露程度或数量及其相互亲和力可以解释为什么PLP1中的各种致病点突变会导致不同程度的过早寡聚。TMD螺旋与内质网和质膜之间的疏水性不匹配可能在WF PLP1的延迟聚合中起作用[58]。而某些PLP1点突变通过改变肽链的疏水性,最终影响PLP1寡聚化的速度[59]。c.某些特定的位点,例如PLP1的细胞外表面内质网管腔环包含4个半胱氨酸残基,被认为在成熟蛋白质中形成分子内二硫键,错误折叠的蛋白质有形成二硫键聚集体的趋势或形成了异常的分子间二硫键而致病[60]。在PLP1的4个螺旋内半胱氨酸中,3种进化中高度保守的C24、C32和C34在蛋白质折叠过程中可能是关键的螺旋-螺旋相互作用所必需的[61]。
内质网中形成的异常寡聚体对少突胶质细胞的存活、髓鞘分子结构的形成等产生致病作用。a.对少突胶质细胞的影响,一种解释是突变体蛋白质合成的速度足够快,足以压倒内质网降解机制,使大量突变体PLP1积聚并损害细胞功能;另一种可能是突变PLP1的一种特殊性质或功能是其毒性的原因。PLP1突变体的表达,已经被证明会不仅导致成熟少突胶质细胞存活率的严重降低且对细胞活力产生极大的负面影响[62]。b.由于正常情况下内质网介导的降解作用(ER-asociated degradation,ERAD)无法处置错误折叠的蛋白质,钙黏连蛋白Calnexin与PLP1突变体相互作用并阻止其降解,从而触发了一些病理过程。错误折叠的PLP1蛋白与钙黏蛋白的稳定且长时间的相互作用,PLP1被保留在ER中[63]。c.PLP1/DM20突变蛋白的积累会激活未折叠蛋白反应(unfolded protein response,UPR),这是一种将错误折叠蛋白的ER积累与核转录抑制相耦合的反馈信号通路[64]。此外,UPR激活可能是将未折叠蛋白的ER积累与凋亡细胞死亡联系起来的常见途径之一[65]。UPR由3个信号通路组成,这些信号通路交互响应错误折叠的蛋白质在ER中积累所引起的ER应激,每个途径均由激活转录因子6(activating transcription,ATF6)、肌醇的酶1(inositol-requiring enzyme,IRE1)和蛋白激酶R样ER激酶(protein kinase R-like ER kinase,PERK)应激传感器启动[66]。在不受压力的条件下,ER伴侣葡萄糖调节蛋白78(ER chaperone glucose-regulated protein 78,GRP78),通过与ER压力传感器ATF6、IRE1和PERK结合而负调控UPR信号通路[67]。当未折叠/错误折叠的蛋白质积聚在ER中时,GRP78与未折叠/错误折叠的蛋白质结合,这导致与ER压力传感器分离,从而启动UPR。ATF6和IRE1-XBP1轴可促进GRP78的表达,从而促进ER蛋白的正确折叠或组装并防止其聚集提高细胞存活率[66,68]。但是,当内质网应激压倒了这种内在质量控制的能力时,C/EBP同源蛋白(CHOP)上调,引发细胞凋亡。PERK途径触发UPR的促凋亡机制,在PMD中大量少突胶质细胞死亡的细胞发病机制中起着核心作用[64,69]。d.突变的PLP1蛋白在ER中积累,不能进入细胞内物质的循环并到达细胞质膜上[70],部分较严重的突变体(msd-A243V)甚至导致高尔基体的碎片化[69],最终细胞膜上髓鞘形成障碍(图4),PMD患者临床症状表现出来。基于以上机制,本团队通过SD-SIM示踪PLP1在COS7细胞内的分布,提出临床上引起经典型表型的突变导致PLP在ER中潴留,而PLP1的剪接体DM20能够表达在细胞表面,相对引起先天型表型的突变则导致PLP1及DM20同时在内质网中潴留[70-71]。此外,本研究表明活细胞结构光成像显微镜能从细胞表型层面预测对应表型严重程度,且相同表型间存在相似的潜在病理机制。导致最严重的细胞表型突变体能够导致PLP1在ER中的潴留已被得到证实,而PLP1的储留进一步导致ER应激和少突胶质细胞死亡[72-73]。其次,PLP1的中间型突变体可以从内质网中逃逸,但错误地靶向囊泡样结构即溶酶体[70,74]。
需要注意的是,一些引起蛋白质截短的突变,如无义及移码突变,很可能是因为通过激活无义介导mRNA降解(nonsense-mediated mRNA decay,NMD)途径,临床上导致相对温和的表型[75]。而在可能改变基因剪接模式的内含子中也发现了点突变[44,76],由此产生的突变体mRNA可能导致外显子跳跃等,相关的表型从严重PMD到轻度SPG2不等,可能取决于突变mRNA的处理方式,特定突变相关的严重程度由其对蛋白质构象和错误折叠的影响来定义[77]。携带框内缺失或插入的突变体,可能导致蛋白质错误折叠,从而导致严重的表型[78]。相反,携带移码的mRNA提前产生终止密码子,可能激活NMD[10,75]。其他一些影响剪接因子的突变,可能导致mRNA不稳定和降解,也可能导致空等位基因,从而导致相对温和的表型。这一事实表明,PLP1点突变导致的严重髓鞘病变可能不是由于缺乏功能蛋白,而是来自突变蛋白的细胞毒性作用[79]。然而,对每个突变为什么表现出的不同轻重表型的实际解释可能更为复杂,相关机理有待进一步阐述。
5.2.2PLP1重复突变的细胞学致病机制
PLP1重复突变是PMD最常见的遗传学病因,重复片段从50 kb~33.5 Mb[32],对于小于1 Mb的重复片段,PLP1可能是造成PMD病理征的唯一基因。最常见的重复片段大小为200~500 kb[80]。导致PLP1重复和缺失的基因组重排的分子机制仍然未知。但在大多数基因组疾病中,引起疾病的基因组重排似乎是由同一染色体中基因LCR之间的非等位基因同源重组(non-allelic homologous recombination,NAHR)介导的[81],但NAHR似乎未在PLP1重复突变相关的PMD中检测到。涉及PLP1的基因组重排被归类为非复发性重排,其中重排的基因组片段在无关患者中具有独特的大小和基因组含量,但都包含PLP1[82]。对PLP1重复和缺失的断点连接的分析表明,重复突变涉及了不同的机制,例如非同源末端连接(non-homologous end joining,NHEJ)和 基 于 复 制 的 机 制(replication-baed mechanism,RBM)包括断裂诱导的复制、微同源介导的断裂诱导的复制、叉停滞和模板切换[83]。RBM是非复发性基因组重排的基础,可导致许多基因组疾病[84]。在PLP1基因座处产生的基因组重排的特征在于复杂的基因组重排,该重排由两个以上的断裂点和多个新生的重复片段穿插并连接在一起。从临床角度来看,此类RBM还可引起PLP1三倍重复,从而导致比重复突变更为严重的表型[85]。同样,在已被用作PLP1过表达模型的Plp1转基因(Tg)小鼠中,具有较高转基因拷贝数的小鼠表现出更严重的表型[86]。具有较低拷贝数的Tg小鼠表现出较轻的髓鞘化低下表型,但却显示了轴突变性,这表明异常量的PLP1也影响了小鼠的轴突维持[87]。而在能够完美契合PLP1重复突变临床表性的Plp1小鼠中,发现了类似的轴突变性与缓慢进行的脱髓鞘并存,而不是典型的髓鞘化低下[88]。而具有PLP1重复的人类在白质中显示出明显的髓鞘过少,由此人类对PLP1基因剂量的敏感性被提出,且阈值可能低于小鼠。而三倍重复患者表现出更重的表型说明PLP1重复突变引起的细胞毒性是剂量依赖性的[84]。
相对于基因敲除小鼠具有明显的轻度表型,PLP1的错义突变或基因剂量增加会导致PMD的严重形式[24]。因此,PLP1致病机制更有可能是毒性物质的“功能获得”(gain of function,GOF)的结果,而不是功能蛋白的丧失(loss of function,LOF)。但是,对于PLP1重复的疾病机制,毒性“功能获得”的分子性质仍然不清楚。有研究表明PLP1过表达会导致少突胶质细胞和髓磷脂的发育停止[86]。但是,这些研究并未显示出抑制的髓鞘发育是PLP1表达的直接作用,还是由于PLP1异常蓄积引起的继发作用。值得注意的是,Plp1-E1和Plp1-E2作为人类中保守的少突胶质细胞最强且特异的增强子,其调节剂Myrf作用于Plp1-E1和Plp1-E2,可以促进PLP1的表达[89]。
PLP1重复增加了PLP1的表达水平,过表达的PLP1与胆固醇积累在少突胶质细胞内体-溶酶体系统中。生理状态下,PLP1通过细胞膜、内质网、高尔基复合体、溶酶体及线粒体等细胞器互作网络(organelle interaction network,OIN)发挥正常的生理功能:PLP1在内质网翻译加工,接下来转运到高尔基体进行分选,通过囊泡分泌到达细胞膜,随后回到晚期内体/溶酶体中。晚期内体/溶酶体是PLP1的“动态调节池”,可以将其储存的PLP1通过囊泡转运到达细胞膜,最终形成髓鞘[46,50]。过表达的PLP1在生物合成运输过程中不与“脂质筏”相关,但在内体系统中形成“筏聚集体”。由于这些筏聚集体干扰了筏膜的运输,因此进一步可能损害髓鞘形成过程(图4)。与此同时,PLP1的过表达导致晚期内体/溶酶体中胆固醇的积聚,晚期内体/溶酶体循环和/或降解髓鞘膜的能力也可能受到干扰,从而诱发或加速疾病进程。此外,在少突胶质细胞的筏中发现了启动髓鞘形成所需的重要信号分子,例如Fyn[90]。在缺乏Fyn的转基因小鼠中,髓鞘形成明显减少,少突胶质细胞的形态分化需要激活Fyn酪氨酸激酶[91],因此推测由PLP1过表达引起筏膜运输受损会破坏Fyn信号传导。
与PLP1点突变不同,PLP1重复突变并不是通过UPR作用对细胞产生毒性作用。在Plp1基因低拷贝数重复的小鼠(Plptg小鼠)中,几乎无法检测到ER中PLP1的异常积累和随后的UPR[92]。而凋亡诱导因子(apoptosis-inducing factor,AIF)在Plp1Tg小鼠细胞核中的水平是Plpjp(jimpy)小鼠的1/3~1/4,AIF水平在细胞核对于介导的细胞死亡途径显著上调[93]。由此,PLP1重复突变产生的细胞毒性作用与PLP1点突变不同。Hüttemann等[92]提出,Plp1tg小鼠表现明显的线粒体功能障碍,表现为氧化还原失衡及ATP产生有关的几种酶被破坏,例如来自Krebs循环和氧化磷酸化的糖酵解酶、肌酸激酶等。而相关的线粒体功能障碍与Mia40/Erv1通路中PLP1中某些特定的半胱氨酸基序例如CX3C和/或CX9C插入线粒体内膜有关[92,94]。综上所述,PLP1重复突变可能通过影响线粒体的功能对细胞产生毒性作用。本研究团队从PLP1重复突变患儿线粒体形态出发,提出PLP1重复突变引起了远核侧内质网“片状化”,内质网-线粒体之间线粒体相关膜(MAM)结构改变,使得线粒体碎片化,从而产生细胞毒性导致线粒体呼吸功能障碍而致病[95]。
5.2.3 无PLP1突变的细胞学致病机制
无PLP1突变的患者都表现出独特的临床表现[81,96],部分患者被诊断为SPG2,尽管这部分患者表型较轻,但他们在之后的生活中常常表现出运动功能的缓慢进行性恶化。具有无PLP1突变的患者通常表现出轻度的外周脱髓鞘性神经病,这在具有其他类型突变的患者中未观察到。电子显微镜结果显示,该类患者少量PLP1位于周围髓鞘,并且PLP1的缺乏,而非PLP1氨基酸改变或过表达会引起周围神经脱髓鞘。PMD和周围神经病患者的研究结果表明,CD2中的PLP1特异性残基是正常外周神经功能所必需的,缺失了这35个氨基酸结构域的突变会导致这种独特的表型[76]。类似的较轻度表型与缺乏PLP1的小鼠的表型和组织学发现一致。无Plp1的小鼠是可以存活的,并且在其CNS中虽然能形成髓鞘,但缺乏PLP1的髓磷脂是脆弱的且压实不完全[24,97]。此外,这些小鼠的轴突肿胀和变性缓慢进行,并在老年时变得明显[98]。起始密码子(即第1个甲硫氨酸)突变或过早产生终止密码子(premature termination codons,PTCs,上游外显子中的无义和移码突变)点突变也可导致类似无PLP1突变。引起PTC的突变可能触发了NMD,这是一种细胞监测系统,用于特异性检测上游外显子中带有PTC的mRNA,以在翻译前对其进行降解[99]。
5.2.4 女性PMD患者中PLP1的致病机制
杂合子PLP1突变的患者表现与经典PMD不同的临床表型[81]。通常神经系统症状要比其他类型的突变温和得多。这些患者通常保持行走的能力,并且他们的认知障碍是轻度的。一些人被诊断出患有SPG2,但为青春期发作的运动疾病,且迟发性或早发性神经学表现的潜在机制可能不同,但两者均可能与X染色体的随机失活有关。X染色体随机失活的结果是杂合的雌性个体具有两个不同的少突胶质前体细胞群,仅表达正常PLP1等位基因的细胞和仅表达突变型PLP1等位基因的细胞。严重的PLP1突变可能影响少突胶质细胞的分化,并导致随后的正常PLP1等位基因失活的少突胶质细胞凋亡。另一方面,具有表达轻度PLP1突变的少突胶质细胞的杂合子可能会通过胚胎发育而存活并形成髓磷脂,从而在成熟的髓磷脂中留下嵌合体细胞群。由于突变的髓磷脂可能是脆弱的或不稳定的,并且可能会降解,因此携带者女性可能会发生随后的脱髓鞘和晚期发病的临床表现[100]。
6 PMD的治疗及预后
截至目前,PMD并无特殊的治疗方法,诊断明确的患者可采取对症治疗、康复训练等。相关研究包括化学或生物制剂的补充及基因治疗两种。
Saher等[101]通过向PLP1转基因小鼠喂食富含胆固醇的饮食,细胞内PLP1的积累能得到改善,少突胶质细胞的数量增加,炎症和神经胶质细胞减少,髓磷脂含量增加,运动缺陷得到改善。吡拉西坦在msd小鼠中被证明可通过增加PLP1的膜定位和降低内质网的应激,但并不能改善其生存率[102]。此外,近年来随着诱导多功能干细胞(induced pleuripotent stem cells,iPSCs)技术及高通量筛选等方法应用于临床研究中,一些小分子物质被证明对PMD临床症状具有改善作用。研究者通过将人类神经干细胞移植到PMD小鼠和患者脑中,发现该疗法在两者体内均产生了大量的功能性髓磷脂且可以延长小鼠的存活期,提示这种疗法可能应用于PMD治疗[103-104]。Emborg等[105]研究表明,在帕金森病猴模型中,移植的自体iPSC衍生的神经祖细胞可分化为神经元、星形胶质细胞和少突胶质细胞。同时研究者们在iPSCs中利用高通量筛选的方法提示,铁螯合剂及一些化合物如Ro25-6981可以挽救少突胶质细胞中铁剂诱导的细胞凋亡和髓鞘形成障碍[51,106]。其次,利用基因疗法在PLP1过表达的少突胶质细胞中,Karim等[107]发现,转染PLP1小分子干扰RNA敲低PLP1可以显著提高MBP阳性髓鞘的形成。Miyamoto等[108]阐明,过表达PLP1对应的原代少突胶质细胞不具有形态分化能力,而其同源siRNA或化学抑制剂对丝裂原活化蛋白激酶/细胞外信号调节蛋白激酶(MAPK/ERK)的抑制逆转它们的未分化表型,从而改善髓鞘形成和运动功能的损伤。腺相关病毒(adenovirus associated virus,AAV)介导的基因特异性抑制可以通过校正基因产物中的定量畸变来作为PMD的潜在治疗方法[109]。向出生后的jimpy小鼠施用单剂量的Plp1靶向反义寡核苷酸(antisense oligonucleotides,ASO)可完全恢复小鼠少突胶质细胞数量、增加髓鞘形成、改善运动性能、使呼吸功能正常化并延长其8个月的寿命[110],这些结果为基因治疗髓鞘疾病建立新的药物模式。
PMD属于髓鞘形成不良性脑白质营养不良的一种,不同的表型预后存在差异。先天型PMD进展快,通常在病程前10年左右迅速进展甚至发生早期死亡。而经典型患者临床症状在病程前10年缓慢进展,可存活至60~70岁。眼震等临床症状可在1岁左右得到缓解,大多数患者在10~12岁之间会逐渐获得运动发育,但是运动水平因患者而异。达到最高能力后,通常会在10~20岁后出现缓慢的恶化,并伴有皮质萎缩等。
为减轻疾病负担对患者家庭产生的影响,建议临床及遗传学诊断明确的PMD患者家系在生育下一胎时要进行遗传咨询,以预防患者家庭中再生出同样患者。
7 小 结
PMD是髓鞘形成低下性脑白质营养不良最具代表性的一种,其典型临床表现包括眼震、肌张力低下及智力运动发育落后等。根据临床症状的轻重,PMD可分为先天型、中间型及经典型3种。先天型起病早,病程进展快,相对的经典型起病较晚,病程进展缓慢。目前针对PLP1突变致病的细胞分子学机制尚未完全阐明,已知PLP1点突变主要引起PLP1错误折叠,错误折叠的蛋白质未能完全转运出内质网而进一步影响髓鞘形成。而PLP1重复突变则可能为过表达的PLP1影响OIN等对细胞产生毒性作用,对PMD患者OIN紊乱机制的探索为阐明PLP1突变致病的细胞分子学机制提供了新思路。
