高效液相色谱-串联质谱法检测水产品中 庆大霉素C组分残留量
2022-11-21杨家锋梁芹芹
◎ 杨家锋,郑 丹,汪 杰,梁芹芹,蒋 海
(宁波市海洋与渔业研究院,浙江 宁波 315010)
庆大霉素在畜牧业及渔业养殖中应用广泛,但同时也带来了药物残留问题。庆大霉素及其残留对人、畜均有明显的毒副作用,长期摄入能诱发急性肾功能衰竭、中毒性耳聋、失语、瘫痪和休克等[2]。
庆大霉素的理化测定方法相当复杂,且灵敏度和通用性较差。因此应从多方面综合考虑庆大霉素的残留测定方法,本方法的主要改进措施有以下几方面。①检测过程尽量避免使用玻璃器具,可采用聚丙烯塑料管,进样瓶亦采用塑料材质衬管等。②提取液为磷酸盐缓冲加三氯乙酸,添加三氯乙酸既可有效去除蛋白质又能提高药物的回收率。③采用MCX混合阳离子交换柱净化浓缩提取液。④使用C18分离柱,流动相中加入五氟丙酸离子对试剂,有利于改善峰型和提高分离效果。⑤使用三重四极杆质谱配备电喷雾离子源,采用ESI+和MRM模式,提高检测选择性和灵敏度,克服了仅以保留时间作为定性依据的缺陷。
1 材料与方法
1.1 试剂
甲醇,乙腈:色谱纯,美国西格玛公司;七氟丁酸,五氟丙酸,三氟乙酸:色谱纯,美国ACROS公司;磷酸氢二钠:分析纯,国产;0.01 mol·L-1磷酸盐缓冲溶液:称取1.36 g磷酸二氢钾,量取30 mL三氯乙酸,用水溶解并定容至1 000 mL;磷酸二氢钾:分析纯,国产;氢氧化钠:分析纯,国产;1.0 mol·L-1氢氧化钠溶液:称取20.0 g氢氧化钠,用水溶解并定容至500 mL;0.02 mol·L-1磷酸盐溶液:称取3.58 g十二水合磷酸氢二钠,用水溶解并定容至500 mL,调整pH=7.0;氨水甲醇溶液:氨水+甲醇=10+90,v/v;1.0‰五氟丙酸溶液:称取1.0 mL五氟丙酸,用水溶解并定容至1 000 mL;庆大霉素:纯度≥98%, CAS No. 1405-41-0,Dr. Ehrenstorfer,美国;标准储备溶液:1.0 mg·mL-1,称取10.0 mg庆大霉素标准品,用水溶解并定容至10 mL,-18 ℃冷冻保存于塑料管中;标准使用液:准确吸取标准储备液,用水稀释配成 1 000 ng·mL-1溶液,现用现配;超纯水:符合分析实验室一级用水规格。
1.2 仪器设备
高效液湘色谱-串联质谱仪,waters2695-QUTTRO MICRO,美国;分析天平,Sartorius BP310S,德国;均质器,FLUKO,上海;氮吹仪,HGC-24A;旋涡混合器,MS2 Minishaker,广州;离心机,DL-8M,上海离心机研究所;真空旋转蒸发仪,BÜCHI R215瑞士;MCX阳离子固相交换柱,Waters,美国;固相萃取装置:HENGAO T&D,HL-02D,巴西;移液枪:上海雷勃;超纯水仪:美国Millipour公司。
1.3 样品处理
1.3.1 制样
取水产品可食部分,切成不大于0.5 cm×0.5 cm× 0.5 cm的小块,充分匀浆,储存于塑料瓶中,密封冷冻(-18 ℃)保藏,备用。
墙夼水库总库容 3.28亿 m3,兴利库容0.85亿 m3,死库容 0.11亿 m3,正常蓄水位98.50m。水库工程防洪标准按百年一遇设计,万年一遇校核。东、西库设计洪水位分别为103.02m、103.16m,校核洪水位分别为106.50 m、106.57m。东西两库共设东库溢洪闸1座,遭遇大洪水时,西库水位高于东库,洪水通过连通沟进入东库,由东库溢洪道泄出。
1.3.2 提取
称取样品(5.0±0.01)g置于50 mL离心管中,加入15 mL磷酸盐缓冲溶液,振荡10 min,设置 4 000 r·min-1转速,离心15 min,上清液过滤后收集。残渣再用15 mL磷酸盐缓冲溶液重复提取1次, 4 000 r·min-1离心15 min后合并上清液滤液,用氢氧化钠溶液调pH值为7.0~7.2,4 000 r·min-1离心 5 min后上清液过固相萃取柱。
1.3.3 净化
MCX柱预处理:将MCX柱安装到固相萃取装置上,过提取液前先依次用5 mL甲醇,3 mL水,5 mL磷酸盐缓冲溶液活化。提取液缓慢过已活化的MCX柱,再依次用2 mL磷酸盐缓冲溶液和5 mL水洗杂质,待柱子干后以3 mL氨水甲醇溶液洗脱,收集洗脱液,氮气吹干。精确量取1.0 mL流动相溶解残留物,过 0.22 µm滤膜,待测。
1.4 标准工作溶液
吸取适量标准使用溶液(1.000 µg·mL-1)用水配制成序列浓度为10.0~1 000.0 ng·mL-1的标准工作溶液。
1.5 标准曲线制作
设定优化后仪器条件,将标准工作液上机测定,以庆大霉素各组分测得结果的峰面积或峰高为纵坐标,以标准工作液的浓度为横坐标绘制标准曲线。
1.6 仪器条件
1.6.1 色谱条件
色谱柱:C18,100 mm × 2.1 mm,5 µm(i.d.),或其他性能相当的色谱柱;柱温:室温;进样量: 30 µL;流动相:0.1%五氟丙酸酸溶液+乙腈=70+30。
1.6.2 质谱条件
离子化模式:大气压电喷雾离子源,ESI+;毛细管电压:2.9 kV;锥孔电压:25 V;脱溶剂气流速:450 L·h-1;锥孔气流速:50 L·h-1;离子传输毛细管温度:350 ℃;离子源温度:110 ℃;碰撞气压力:氩气, 2.5 mTorr;扫描模式:多离子反应监测模式(MRM),选择反应监测母离子、碎片离子和碰撞能量见表1。

表1 选择反应监测母离子、碎片离子和碰撞能量表
1.7 结果计算
样品中庆大霉素残留量按式(1)计算,计算结果需扣除空白值。

式中:Xi为样品中庆大霉素C组分的含量,μg·kg-1;Ci为样品制备液中庆大霉素各组分的浓度,ng·mL-1;V为最终定容体积,mL;m为样品质量,g。
2 结果与分析
2.1 庆大霉素C组分的质谱特征
用水配制1 000 ng·mL-1的标准溶液用蠕动泵进样方式直接导入ESI源,首先采用一级全扫描质谱方式,获得待测物的准分子离子峰[M+H]+,然后对准分子离子峰及其碎片离子,采用Daughter scan质谱模式进行二级质谱分析,以获取相应的多级质谱信息。
本实验中的庆大霉素(图1)由3个环组成,A环和C环为取代的氨基葡萄糖,B环为取代的脱氧链霉胺(中心单元)[3]。庆大霉素的结构中含有多个碱性中心,其裂解机制是由正电荷引发的i过程,失去的都是中性分子碎片,见图2,表2。

图1 庆大霉素分子结构图

图2 庆大霉素碎片离子裂解图

表2 庆大霉素碎片离子裂解表
在一级全扫描质谱条件下,分别获得4种结构相似的准分子离子峰m/z450,m/z464和m/z478(庆大霉素4种组分)(图3)。为进一步了解该类抗生素的质谱断裂规律,分别对各准分子离子峰进行多级质谱分析,获得相应的碎片离子(图4、图5、图6)。

图3 庆大霉素母离子质谱图

图4 庆大霉素C1a子离子质谱图
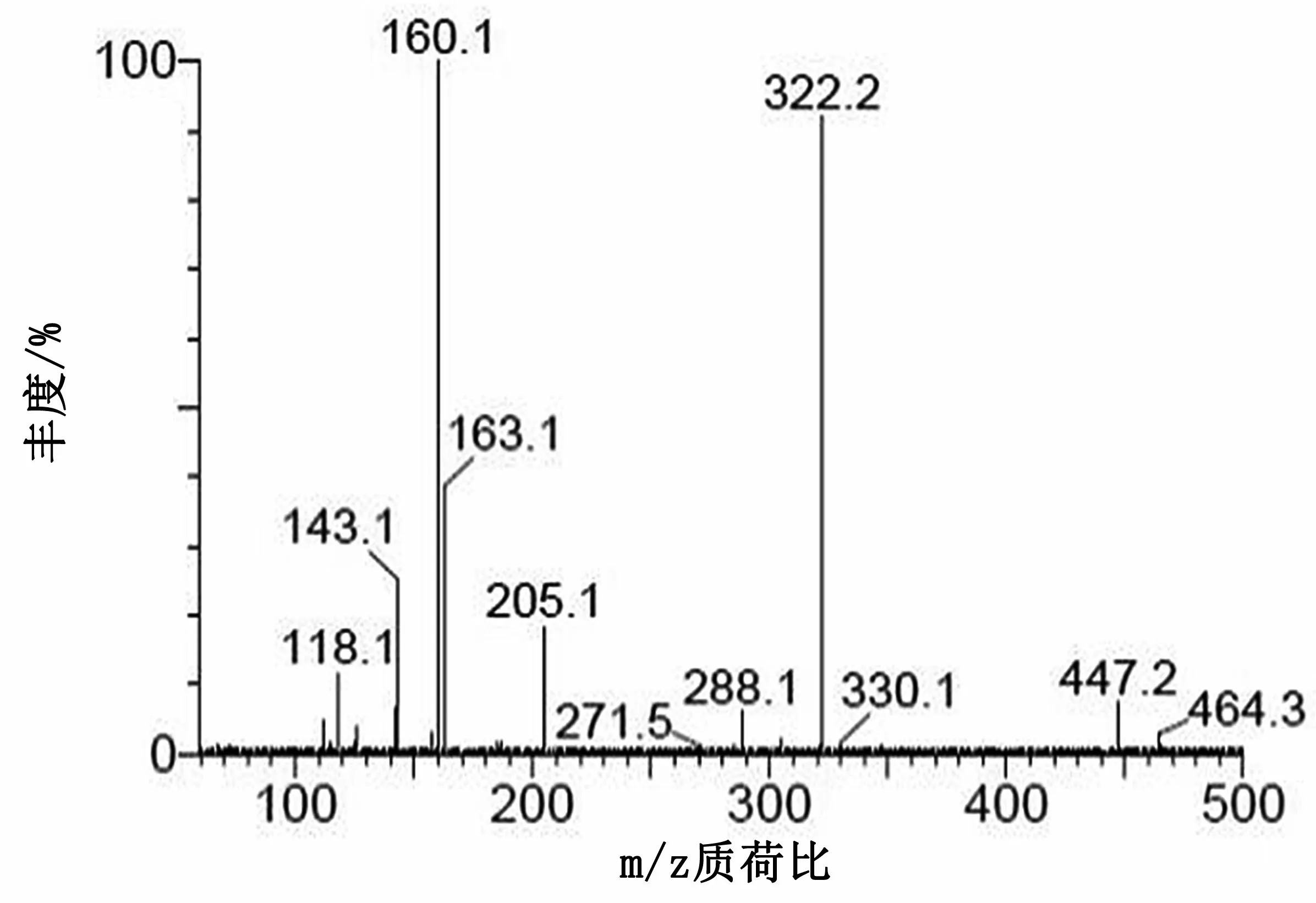
图5 庆大霉素C2+C2a子离子质谱图

图6 庆大霉素C1子离子质谱图
在二级质谱分析中,庆大霉素C组分由4种(C1,C1a和C2+C2a)组成,对应3个准分子离子峰,分别选择m/z478,m/z450和m/z464进行二级质谱分析,均形成同样的碎片离子m/z322,进一步证明本实验中的庆大霉素在二级质谱分析中首先脱去C环。加大碰撞能进行二级质谱分析,m/z322可生成m/z160(脱去B环)、m/z163(脱去A环)和m/z205等碎片离子。m/z160可进一步脱水分别生成m/z142碎片离子。m/z205推测是A环断开,然后脱水形成的碎片离子。
2.2 质谱条件优化
1 μg·mL-1的庆大霉素标准溶液以蠕动泵方式进 样,流速为20 μL·min-1,在ESI+模式下进行一级质谱图扫描,以4种组分的[MH]+(C1,C1a和C2+C2a组成对应3个准分子离子峰分别为m/z478,m/z450和m/z464)峰灵敏度为依据,优化质谱调谐文件包括毛细管电压、离子源温度、锥孔电压、脱溶剂气等参数,见1.6.2。分别以m/z450,m/z478和m/z464作为母离子,对其子离子进行扫描,选取丰度最强的子离子m/z450>322,m/z478>322和m/z464>322为 定量离子,次强的子离子m/z450>160,m/z478>157和m/z464>160作为定性离子;以子离子强度为依据优化碰撞能量等的质谱条件见表1。
2.3 色谱条件的选择
本实验以三氟乙酸、五氟丙酸、七氟丁酸为离子对试剂进行试验,浓度设0.5‰、1.0‰、1.5‰3个水平,以乙腈与离子对试剂溶液多组比例为流动相进行比较实验。结果表明,用1.0‰五氟丙酸与乙腈比例为70∶30时,色谱峰的峰型、分离度和灵敏度都到达最优状态,保留时间为2.91 min,标准溶液5 ng·mL-1时,定量子离子峰的信噪比大于15。随着流动相中乙腈的浓度减少,目标物质保留时间延长。当乙腈含量为20%,保留时间为4.57 min,但灵敏度降低。以七氟丁酸为离子对试剂对庆大霉素的分离和提高色谱柱的保留有不错的表现,在乙腈含量为20%,保留时间为7.21 min,但灵敏度比使用五氟丙酸时低。以三氟乙酸为离子对试剂,庆大霉素的色谱峰峰宽较大且有明显峰拖尾。因此,选取1.0‰五氟丙酸+乙腈=70+30为流动相。
流动相中五氟丙酸浓度分别为0.5‰、1.0‰时,对保留时间及分离度的影响不大。流动相pH值在2.5~3.5内,容量因子改变不大,但pH值小于2.5或pH值大于4.0时,流动相容量因子急剧降低。在pH值为2.5~3.5时,五氟丙酸带负电荷,而庆大霉素各组分分子带正电荷,两者形成离子对化合物,在反相色谱柱上被保留。1.0‰五氟丙酸的pH值在2.5~3.0,因此一般无需调节pH值。但当流动相pH值偏小时,庆大霉素各组分会出现双峰现象。
在流动相为1.0‰五氟丙酸+乙腈=70+30时,比较了waters的Atlantis、Sunfire、Xterra、Symmetry以及资生堂的MGⅢ、CAPCELL-Pak混合柱等色谱柱对庆大霉素的出峰效果。结果表明,Atlantis C18的出峰效果最佳。
2.4 样品处理条件优化
2.4.1 提取溶剂的确定
本方法以乙腈、二氯甲烷和乙酸乙酯有机溶剂以及磷酸盐(KH2PO4)、高氯酸、三氯乙酸、三氟乙酸、氢氧化钠作为提取溶剂进行回收率试验。试验结果表明用乙腈、二氯甲烷和乙酸乙酯有机溶剂提取试样中庆大霉素药物时,回收率很低,而且用乙酸乙酯提取时甚至没有回收到目标物,该结果与庆大霉素的溶解性有关。氢氧化钠溶液虽然能较好地提取试样中庆大霉素,但在提取组织试样时易形成乳胶状态,不利于后续步骤的进行。2%~5%三氯乙酸或高氯酸既可有效脱蛋白质,也可以溶解庆大霉素提高回收率。三氟乙酸脱蛋白效果稍劣于三氯乙酸。因此,通过多组实验比较后决定用磷酸盐和三氯乙酸的混合溶液提取组织中庆大霉素,回收率高达89%~103%。DAVID等[4]采用30%的三氯乙酸提取血浆和尿中的庆大霉素,回收率为92%~107%,与本文结果较一致。
2.4.2 试样的净化
由于试样中庆大霉素的提取剂为磷酸盐和三氯乙酸的混合溶液,试样提取液不易用旋转蒸发的方式浓缩,固相萃取选择吸附便是一种理想的浓缩净化方法。本试验以HLB、C18、MCX、SAX、WAX、PRS柱为净化固相萃取柱,实验结果表明C18、MCX、SCX对庆大霉素都有较好的保留,其中又以MCX结果最优,过柱回收率达97%以上,因此确定以MCX为本方法净化固相萃取柱。过柱洗脱液为甲醇氨水溶液,在洗脱过程中第1 mL可将柱中95.5%的庆大霉素洗下,第2 mL又能洗下3.5%,因此确定洗脱液为3 mL。洗脱液氮吹至近干,准确量取1.0 mL流动相定容。ELIANGIRINGA等[5]使用CBA弱离子交换柱除去蛋白质后测定血清中的庆大霉素,回收率为78%~93%。
3 方法评价
3.1 回收率和精密度
以阴性大黄鱼、南美白对虾、梭子蟹为检测试样,每份5.00 g(称准至0.01 g),分别添加庆大霉素,添加水平为分别为5 µg·kg-1、20 µg·kg-1、100 µg·kg-1。检测结果表明,方法平均回收率在71.9%~101.0%,批内、批间变异系数均小于15%。本方法中认定的回收率为70%~110%。
3.2 线性关系和线性范围
配制庆大霉素药物标准的工作曲线浓度系列为 5 µg·L-1、10 µg·L-1、20 µg·L-1、50 µg·L-1、100 µg·L-1、200 µg·L-1、500 µg·L-1和800 µg·L-1,用液相色谱-串联质谱分析,庆大霉素4种成分在5~800 µg·L-1具有很好的线性。庆大霉素4种成分标准溶液工作曲线见图7、图8、图9,线性方程及定量限见表3。

表3 庆大霉素标准溶液工作曲线及方法检出限表

图7 庆大霉素C2+C2a标准溶液工作曲线图

图8 庆大霉素C1标准溶液工作曲线图

图9 庆大霉素C1a标准溶液工作曲线图
3.3 检出限
对阴性大黄鱼、南美白对虾和梭子蟹分别以 5 µg·kg-1加标量进行加标回收试验,试验结果表明回收率都在71.9%以上,信噪比大于15。因此将5 µg·kg-1作为最小检出浓度。
4 结论
本文构建了一种液相色谱-质谱联用法检测水产品中庆大霉素C组分残留量的方法,实验结果表明,庆大霉素4种C组分在0.005~0.800 µg·mL-1,线性相关系数良好,相关系数r>0.999 8;以5 µg·kg-1、20 µg·kg-1、100 µg·kg-13浓度水平进行加标回收实验,回收率在71.9%~101.0%,RSD<15%;当加标量为5 µg·kg-1时4种C组分均具有明显的响应峰,信噪比≥15。该方法适用于水产品中庆大霉素C组分残留量的测定。
