陈化烤烟叶片不同发酵时间微生物多样性及代谢组分析
2022-11-10张文友颜朗赖先军段旺军张义正
张文友,颜朗,赖先军*,段旺军,张义正
陈化烤烟叶片不同发酵时间微生物多样性及代谢组分析
张文友1,颜朗1,赖先军1*,段旺军2,张义正3
1 西昌学院农业科学学院,攀西特色作物研究与利用四川省重点实验室,四川省西昌市安宁镇学府路1号 615013;2 四川中烟工业有限责任公司,成都市龙泉驿区国家成都经济技术开发区成龙大道龙泉段2号 610066;3 四川大学生命科学学院,分子生物学及生物技术四川省重点实验室,四川省成都市一环路南一段24号 610064
【目的】了解不同陈化发酵时间烟叶中微生物菌群与代谢产物的关系,探究多年陈化发酵过程中微生物对烟叶发酵品质的影响。【方法】选取4个陈化发酵时间(陈化0年,1年,4年,7年)的上部烟叶(B2F),采用Illumina高通量测序技术和UPLC-ATOF MS液质联用技术,对不同发酵时间的烟叶表面微生物多样性以及差异代谢物进行分析。【结果】相比其他陈化发酵时间,发酵4年的烟叶中微生物种类最多、多样性最高。在细菌属分类水平上,泛菌属()和假单胞菌属()为发酵4年烟叶的优势细菌。结合OPLS-DA模型以及数据库检索,4个发酵时期两两比较共筛选到173个显著差异代谢物,KEGG富集分析发现差异代谢物参与氨基酸代谢、脂肪酸代谢、类固醇生物合成等多条代谢途径。其中,发酵4年样本中高丰度的差异代谢物主要富集在脂肪酸降解途径。【结论】复烤烟叶在陈化发酵不同时间微生物多样性及代谢物质具有显著差异,上部叶片陈化发酵4年烟叶的内在品质最佳。
复烤烟叶;陈化发酵;微生物多样性;代谢组;烟叶品质
复烤烟叶陈化发酵过程中品质变化的主要原因是烟叶中化合物的分解和转化。例如,不具备香味特性的大分子物质转化为具有香味特性的小分子化合物,从而影响烟叶的香气和吃味品质[1-2]。烟叶微生物可代谢产成酸类、单萜类、酯类和氮杂环类等多种小分子物质或中间产物,从而增加致香化合物生成、转化与积累的效率和速度,因此,探索烟叶发酵过程中微生物群落演替及代谢产物的变化是提高发酵烟叶品质的重要途径[3-4]。
前人对发酵烟叶微生物的变化的研究主要基于微生物分离鉴定的方法,然而多数微生物因无法纯培养而未能被检测[5]。随着高通量测序技术的发展,科研人员能够以更低的成本和更短的时间对复杂菌群进行深入精准的鉴定,解决传统方法存在的不足[6]。本研究利用高通量测序技术,结合代谢组学分析,解析了发酵烟叶中的微生物组和代谢物谱,为进一步提高陈化烟叶品质提供理论依据。
1 材料与方法
1.1 实验材料
供试品种为红花大金元,产自四川省凉山州乌东德镇新马乡,烟叶等级为B2F。各年份贮藏烟叶在入库前均采用近红外检测其内在质量(表1),各指标在不同年份入库烟叶间差异不显著(显著性分析基于SPSS软件LSD最小显著性差异法完成)。烟叶封箱后进入同一仓库的同一贮藏高度储存,仓库位于四川省凉山州德昌县烟叶复烤厂区,库内相对空气湿度控制在(60±2)%,温度控制在(30±5)℃。
表1 各年份烟叶入库前内在成分含量表(等级B2F)
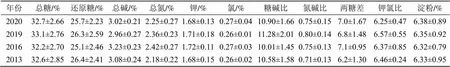
Tab.1 Chemical composition of tobacco leaves in different years before storage (grade B2F)
1.2 实验方法
1.2.1 取样方法
以复烤后片烟封箱入库的时间作为起始发酵时间,分别选取陈化0年(Nt2020)、1年(Nt2019)、4年(Nt2016)和7年(Nt2013)的样品进行取样,将样品随机分为3个生物学重复分装至样品袋内密封,4℃保存。
1.2.2 DNA提取和PCR扩增
利用DNA提取试剂盒(美国MOBIO公司)对发酵烟叶样品中微生物基因组DNA进行抽提,利用琼脂糖凝胶电泳检测抽提的基因组DNA质量,随后稀释至3.5 ng/μL并保存于-20℃条件下用于PCR扩增。使用通用引物338F(5’-ACTCCTACGGGAGGCAG CA-3’)和806R(5’-GGACTACHVGGGTWTCTAAT- 3’)扩增细菌16S rRNA基因的 V3-V4 区域[7]。
1.2.3 文库构建和测序
PCR产物经纯化、等摩尔浓度定量富集和均一化形成测序文库,经文库纯化、质检后,委托北京百迈客生物科技有限公司完成MiSeq测序。
1.2.4 测序数据分析
使用Trimmomatic v0.33软件[8]对Raw Reads进行过滤,Cutadapt 1.9.1软件进行引物序列的识别与去 除[9],Usearch v10软件[10]进行拼接,UCHIME v4.2软件[11]鉴定并去除嵌合体序列,最后对数据进行划分Feature(OTUs、ASVs)、多样性分析、差异分析、相关性分析及功能预测分析。使用R/Bioconductor包edgeR[12]中trimmed mean of M values(TMM)方法对OTU矩阵进行标准化,得到OTU相对丰度矩阵。使用 RDP Classifier 软件(v2.2,置信度阈值0.8)[13]并基于Silva分类学数据库对OTU进行分类学注释。
1.2.5 菌群多样性统计与显著性分析
细菌菌群多样性分析采用实验室自编程序完成。采用R语言edgeR包中glmfit函数进行OTU丰度差异分析并采用stats包中wilcox.test函数进行Wilcoxon秩和检验。使用R中ggplot2包完成文中所有配图。在微生物功能预测中,使用PICRUSt2软件进行物种注释[14],使用IMG微生物基因组数据进行功能信息输出,推测样本的功能基因组成。使用STAMP[15]中的G-TEST检验方法进行两两样本间的显著性差异检验,-value阈值为0.05。使用KEGG数据库[16]完成微生物代谢功能的富集。
1.2.6 代谢物提取和色谱质谱分析
称取50 mg样本,加入1000 μL含有2 μL内标L-2-氯苯丙氨酸(中国阿拉丁公司)的提取液,涡旋混匀后加入瓷珠45 Hz研磨处理10 min,冰水浴超声10 min,-20℃静置1 h。4℃下,12000 r/min离心15 min后,取500 μL上清,在真空浓缩器中干燥提取物,再加入160 μL提取液复溶,涡旋后冰水浴超声10 min,4℃下12000 r/min离心15 min。取120 μL上清于2 mL进样瓶,同时从每个样本各取10 μL混合后上机检测。
使用液质联用系统(超高效液相色谱:Waters UPLC Acquity I-Class PLUS,美国Waters公司;高分辨质谱:Waters UPLC Xevo G2-XS QTof,美国Waters公司)完成色谱质谱分析。正负离子模式扫描下,流动相 A为0.1%甲酸水溶液,B为0.1%甲酸乙腈,进样体积为1 μL,梯度洗脱条件见表2[17]。
表2 流动相梯度洗脱条件

Tab.2 Gradient elution conditions of mobile phase
1.2.7 代谢组数据处理及统计分析
采集的原始数据通过Progenesis QI软件进行峰提取、峰对齐等数据处理操作[18]。基于Progenesis QI软件在线METLIN数据库进行代谢物鉴定,最后以两组间代谢物差异的显著性(-value<0.05,t检验)和OPLS-DA模型的VIP值(VIP>1)为标准筛选获得差异代谢物,并使用MetaboAnalyst结合KEGG数据库进行代谢通路分析[19-20]。
2 结果与分析
2.1 不同陈化发酵时间烟叶感官质量评价
对不同陈化发酵时间烟叶样本的内在质量评价结果表明(表3),Nt2016(发酵4年样本)较其他年份样本烟叶香气质及香气量显著提高,表明烟叶香气质和香气量在陈化4年时间后达到最佳。进一步延长陈化时间,烟叶内含物质进一步分解,烟叶的香气量降低。
表3 陈化后烟叶感官质量评价表(等级B2F)
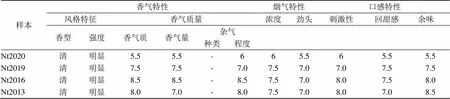
Tab.3 Evaluation of sensory quality of flue-tobacco leaves at different fermentation stages (grade B2F)
注:各指标均分值采用9分制。
Note: The average score of each index adopts the 9-point system.
2.2 不同陈化发酵时间烟叶微生物菌群多样性
对不同发酵时间烟叶菌群物种丰富度和多样性的测定结果表明(图1),随着测序深度增加,稀释曲线基本达到饱和,说明测序数据能够反映发酵烟叶中的微生物多样性。不同发酵时间样本稀释曲线的分布结果表明,Nt2016样本中细菌菌群多样性最高,其次为Nt2013和Nt2019样本,复烤后陈化时间不足1年的Nt2020样本中细菌菌群多样性最低。
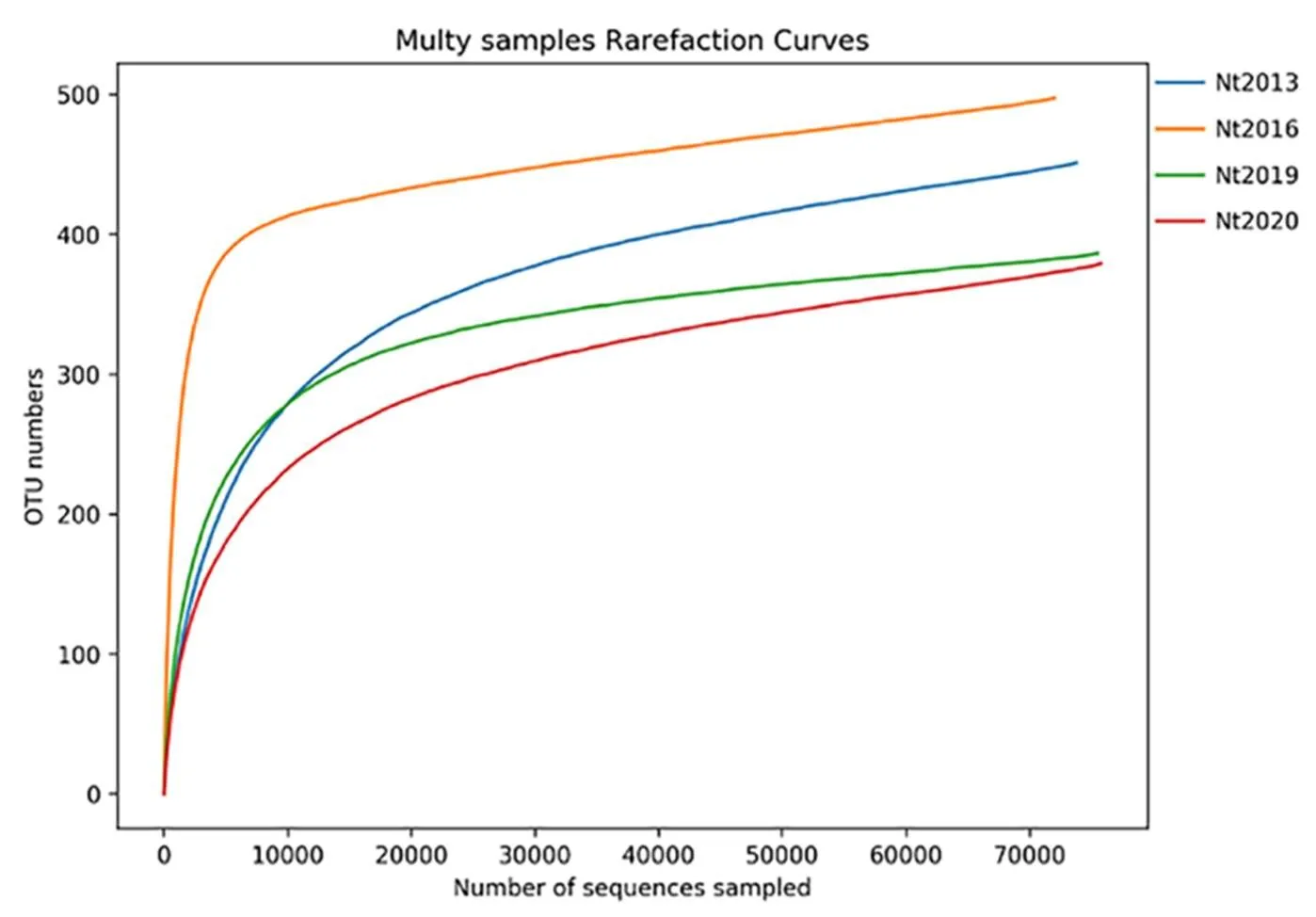
图1 发酵烟叶测序样品稀释曲线
进一步分析每个样本的Coverage覆盖度指数、Chao1丰富度指数、Shannon多样性指数以及Simpson多样性指数。由表4可知,不同发酵时间的烟叶中的Coverage覆盖度指数都大于0.99,表明样品文库中的序列基本都被测出,数据可用于后续分析。α多样性分析表明,随着发酵时间的延长,Chao1指数、ACE指数和Shannon指数先升高后降低。在Nt2016中,物种丰度和多样性达到最高。
表4 不同样本的α多样性指数

Tab.4 α-diversity indices of different samples
进一步构建柱状图展示样本在不同分类水平上的群落结构分布,不同陈化发酵时间烟叶在细菌门水平上的多样性变化情况如图2A所示。在所有发酵时间的烟叶中均发现10个菌门,分别是髌骨细菌门(Patescibacteria),广原菌门(Euryarchaeota),绿弯菌门(Chloroflexi),疣微菌门(Verrucomicrobia),酸杆菌门(Acidobacteria),拟杆菌门(Bacteroidetes),放线菌门(Actinobacteria),厚壁菌门(Firmicutes),变形菌门(Proteobacteria)和蓝细菌门(Cyanbacteria)。其中,蓝细菌门和变形菌门占主导地位,随着陈化发酵时间的延长,蓝细菌门的相对丰度逐渐降低,在Nt2016发酵时间降到最低,而随着发酵时间的延长,其相对丰度有所回升;变形菌门的相对丰度先升高再降低,在Nt2016发酵时间达到峰值。另外,在厚壁菌门、放线菌门和拟杆菌门中,细菌相对丰度均随着陈化发酵时间的延长逐渐增加。
不同发酵时间烟叶中细菌属水平上的多样性变化情况如图2B所示,在所有发酵时间中,占主导作用的优势菌群主要是未培养的含叶绿体细菌(uncultured bacterium Chloroplast),甲基杆菌属细菌(),线粒体中未培养细菌(uncultured bacterium Mitochondria),泛菌属细菌(),鞘脂单胞菌属细菌(),肠杆菌科未培养细菌(uncultured bacterium Enterobacteriaceae),假单胞菌属细菌(),肠道菌科未培养细菌(uncultured bacterium Muribaculaceae),罗尔斯通菌属细菌(),不动杆菌属细菌()。其中,泛菌属细菌在Nt2019发酵时间开始相对丰度逐渐增加,特别是在Nt2016发酵时间丰度激增,而随着陈化发酵时间的进一步延长其相对丰度骤减。

图2 不同发酵时间烟叶细菌在菌门(A)和菌属(B)水平上相对丰度变化
2.3 不同陈化发酵时间烟叶微生物功能分布及差异比较
为了进一步探索Nt2016发酵时间与其他3个发酵时间烟叶中微生物菌群差异,通过微生物基因功能预测及KEGG代谢途径差异分析,确定了Nt2016样本与其他发酵时间样本两两间微生物群落的功能基因在代谢途径上的差异和变化。如图3所示,随着陈化发酵时间的延长,群落样本为适应环境变化发生了代谢功能改变,其丰度变化规律主要可分为两个类型。其一是在整体代谢图谱(Global and overview maps)、能量代谢(Energy metabolism)、辅因子与维生素代谢(Metabolism of cofactors and vitamins)方面,随着发酵时间的延长,相关微生物丰度先显著增加,而在后期发酵4年和7年又显著下降。即在Nt2020与Nt2019比较中,Nt2019中微生物丰度比例显著高于Nt2020,而在Nt2016和Nt2013的相关微生物丰度均显著低于Nt2020。其二是在细胞运动(Cell motility)、信号转导(Signal transduction)和碳水化合物代谢(Carbohydrate metabolism)代谢通路中,随着发酵时间的延长,相关微生物丰度先显著下降,但在发酵4年时间处理的烟叶显著增加,后在发酵7年时间其丰度又有所降低。即在Nt2020与Nt2019比较中,其95%置信度区间内微生物丰度占有较大优势,而在Nt2020与Nt2016的比较中,Nt2016中微生物丰度比例显著高于Nt2020。Nt2020与Nt2013相比,Nt2013中微生物丰度比例也显著高于Nt2020,但95%置信度区间内两者的差异比例小于Nt2016。该结果表明,随着发酵时间的延长,微生物整体代谢活性在不断降低,而在部分代谢途径中,Nt2016发酵时间的相关微生物较为活跃。

注:图中A~C左边所示为不同代谢途径的微生物在两两样本间的丰度比例,中间所示为95%置信度区间内微生物丰度的差异比例,右边为显著性,P<0.05。
2.4 复烤烟叶陈化不同发酵时间代谢组分析
对不同发酵时间烟叶的代谢组分析结果表明(图4),正负离子扫描模式下所有质控样本的变化分布于2STD之内,且正负样本的PCA分布中所有样本QC相对集中、组内差异不明显、组间差异明显、代谢组数据质量高,可以进行后续分析。利用主成分分析(PCA)方法将多维数据降维评估样品的总体差异发现(图4),不同发酵时间的烟叶样品组内聚集,距离较小,而样品组间区分明显,距离较大。该结果一方面表明测定结果重复性较好,同时表明不同发酵时间的烟叶样品具有明显差异,其体系内代谢物发生明显变化。其中,Nt2020与Nt2019组间差异较小,Nt2016与Nt2013组间差异也较小,而在Nt2019与Nt2016间代谢物差异较大,表明其代谢物的主要变化时间应集中在发酵2~4年之间。
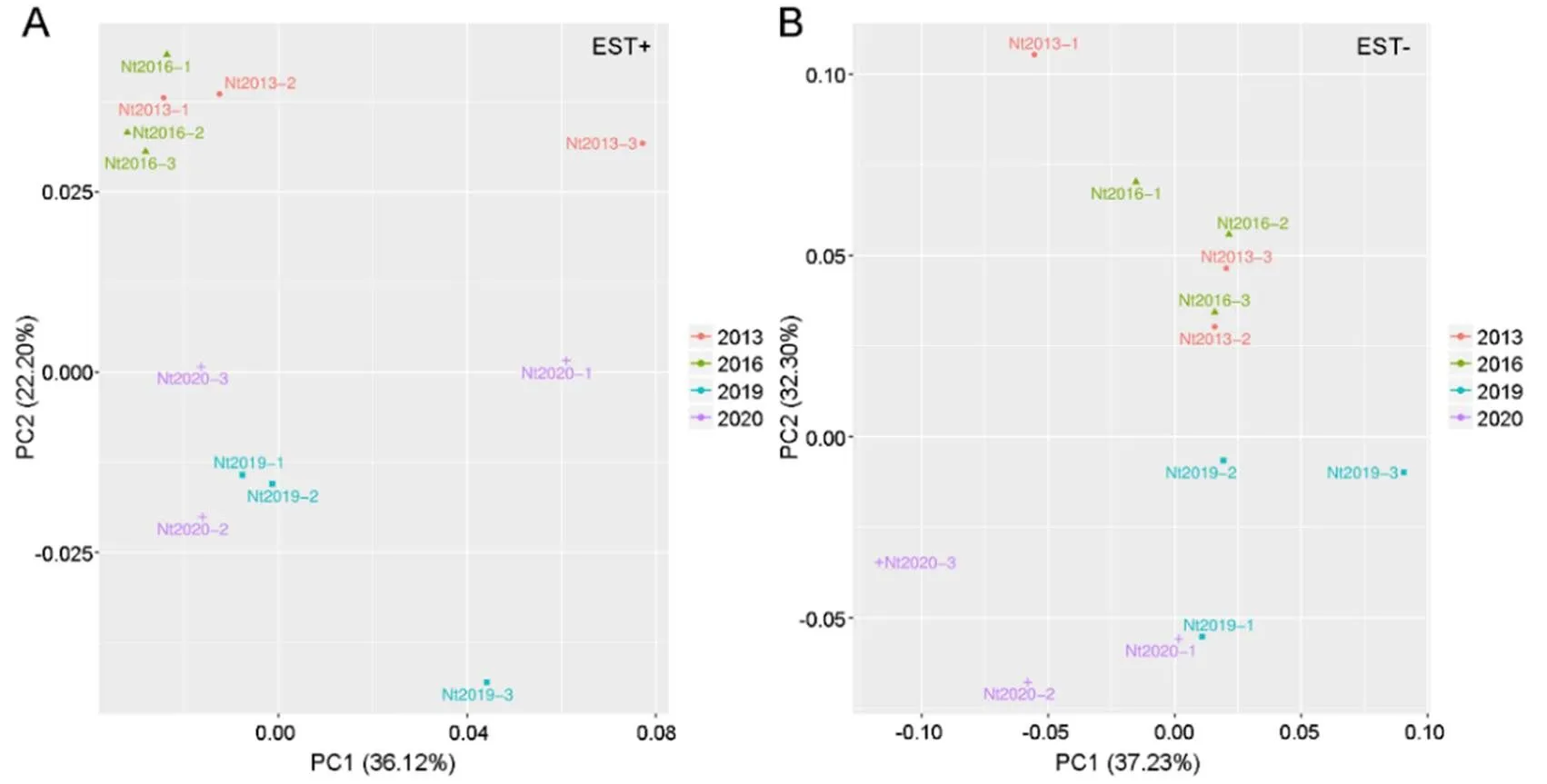
图4 不同发酵时间发酵烟叶的PCA分析图
利用OPLS-DA法[21](orthogonal partial least squares discriminant analysis, OPLS-DA)分析不同发酵时间烟叶两两样本间代谢物的差异,结果如图5所示,正离子模式下,4个发酵时间每两组间分离效果良好。模型可靠性验证表明,6个模型稳定,结果可信度高。

图5 不同发酵时间两组间发酵烟叶OPLS-DA图
基于不同发酵时间两两样本间代谢物丰度经t检验后的值(<0.05)和OPLS-DA模型的VIP值(VIP>1),筛选出6个比较组间共计173个具有显著性差异的代谢物,每组分别产生了15~96个显著差异代谢物,以OPLS-DA模型的VIP值从高到低排列Top5的显著差异代谢物如表5所示。在这些差异代谢物中,玉米烯酮在3个Nt2016参与的比较组中都表现出显著高丰度,1-棕榈酰-2-羟基-丙三基-3-磷酸乙醇胺和肉毒碱代谢物丰度在2个Nt2016参与的比较组中显著增高。另外,在Nt2013参与的比较组中,(E)-2-丁烯基-4-甲基-苏氨酸和β-高脯氨酸的丰度相对于Nt2019和Nt2020样本中的丰度显著增高,表明其在长期发酵过程中不断积累。去甲丙咪嗪和罗布麻苷在Nt2020样本中的丰度显著高于Nt2019和Nt2013样本,表明其在烟叶初期发酵中可能发挥重要作用。
表5 六个比较组中VIP值排名前五的差异代谢物

Tab.5 The top 5 differentially accumulated metabolites with VIP values in 6 comparison groups
续表5

比较组差异代谢物总数VIP值前五的差异代谢物Log2FCPVIP代谢物类型 Nt2016 vs Nt202096玉米烯酮Zearalenone0.978.09E-041.44聚酮类化合物polyketides 肉毒碱Carnitine1.289.34E-041.43氨基酸及其衍生物Amino acids and derivatives 4-羟基-2,3,9-三甲氧紫檀碱4-Hydroxy-2,3,9-trimethoxypterocarpan1.530.93E-031.42紫檀烷类化合物pterocarpan Halaminol A2.781.06E-041.42 5-甲基-2-巯基尿苷5-Methyl-2-thiouridine1.621.14E-031.42核苷类似物Nucleoside Nt2019 vs Nt202052去甲丙咪嗪Desipramine-5.921.84E-041.63 罗布麻苷Cymarin-3.172.41E-041.62类固醇Steroids 麦角灵-1-吡咯甲醛,8-(羟甲基)-10-甲氧基-6-甲基-Ergoline-1-carboxaldehyde,8-(hydroxymethyl)-10-methoxy-6-methyl-0.259.77E-041.60 N-甲基-D-天冬氨酸N-Methyl-D-aspartic acid1.268.48E-041.60氨基酸及其衍生物Amino acids and derivatives 环戊醇胺酯Cyclopentolate0.678.94E-041.59抗胆碱类化合物Anticholinergics
利用clusterProfiler软件选用超几何检验的方法对差异代谢物进行KEGG富集,结果如图6所示,两两比较组中的显著差异代谢物参与的代谢通路有亚麻酸代谢、精氨酸和脯氨酸代谢、色氨酸代谢、脂肪酸生物合成、脂肪酸降解、半乳糖代谢、亚油酸代谢、赖氨酸生物合成、赖氨酸降解、类单萜生物合成、戊糖葡萄糖醛酸转换、磷酸盐代谢、嘧啶代谢、类固醇生物合成、牛磺酸代谢、萜类骨架生物合成。在上述富集的代谢通路中,脂肪酸降解通路在Nt2016样本与其他样本相比较都具有显著差异。另外,类固醇生物合成途径在Nt2020样本与其他样本相比较均有显著差异,表明其可能是烟叶陈化初期发酵的主要代谢途径。
图6 两两样本比较中的差异代谢物及其参与的代谢途径网络图
Fig.6 The network between the differentially accumulated metabolites and enriched pathway in six pair-wise groups.
3 讨论
烟草吸食品质与复烤烟叶发酵过程中微生物的作用密切相关。Li等研究发现,人工发酵2周、4周、6周的发酵烟叶中假单胞菌属、鞘脂单胞菌属、甲基杆菌属、泛菌属为优势菌属,随发酵时间延长,假单胞菌丰度增加,鞘脂单胞菌属和甲基杆菌属丰度降低[22]。龚俊等利用克隆文库法和高通量测序法对烟叶微生物类群的研究发现,假单胞菌属、不动杆菌属和鞘氨醇单胞菌属为主要优势细菌[23]。本研究发现在陈化发酵4年后(Nt2016),烟叶中的微生物丰度达到最大,微生物种类最多,其中,泛菌属和假单胞菌属在该样品中丰度达到最大。以甲基杆菌属为代表的菌属则在新发酵烟叶(Nt2020)中丰度达到最大,并随着发酵时间的延长其丰度逐渐降低。
前人研究表明,假单胞菌具有降解烟碱的能力,且降解烟碱时以烟碱作为唯一碳源和氮源从而成为优势菌属[24]。因此,假单胞菌属在降低烟草吸食危害,改变可燃卷烟的成瘾性中起重要作用。鞘氨醇单胞菌作为从假单胞菌属中分离出来的新菌属,具有独特的脂多糖结构,繁殖能力强,广泛应用于生物大分子的降解方面[25]。有研究表明,经鞘氨醇单胞菌发酵处理后,烟叶淀粉含量显著降低,总糖、还原糖含量增加,同时鞘氨醇单胞菌能够降解烟草中绿原酸及芸香苷等多酚物质,使烟叶感官质量提高[26]。Zhao等证实了泛菌属微生物可以降解烟叶中的类胡萝卜素,并生成巨豆三烯酮、二氢大马酮等香气物质,改善烟草的香气质量[27]。本研究发现,这些与烟叶品质相关的优势菌属在不同发酵时间的烟叶中丰度存在差异,这可能与不同时间的发酵烟叶品质有关。同时,本研究利用PICRUSt方法对鉴别到的微生物进行代谢功能注释,结果表明,随着发酵时间的延长,以能量代谢为主的微生物整体代谢活性在不断降低,而与细胞能动性、信号转导和碳水化合物代谢相关特异性微生物丰度在Nt2016中显著增加,表明陈化发酵4年时烟叶中微生物代谢活性发生显著变化。
本研究通过代谢组学分析鉴定了不同发酵时期烟叶中的差异代谢物。其中,玉米烯酮在Nt2016参与的所有比较组中都表现出显著高丰度。玉米烯酮是常见的霉菌毒素之一,由霉菌产生[28]。早在1933年就有研究发现,在烟叶陈化发酵过程中,细菌和霉菌是烟叶表面主要的两大类群微生物[29]。本研究中Nt2016的发酵烟叶产生高丰度的真菌代谢产物,表明在Nt2016的发酵烟叶中不仅细菌的多样性和代谢活性显著增加,同时也应该存在着高丰度的真菌。另外,在Nt2013与其他样本间的比较组中发现,苏氨酸、脯氨酸、天冬氨酰-精氨酸、丙氨酰-苯丙氨酸这些氨基酸分子在组间差异积累显著。研究表明苯丙氨酸能够降解生成苯甲醛、苯乙醇及苯乙醛,这些物质为烟叶香气物 质[30],因此长期陈化发酵确实有利于香气物质的积累。KEGG富集分析发现这些差异代谢物参与到氨基酸代谢、脂肪酸代谢等代谢合成途径。其中,脂肪酸降解途径在Nt2016的发酵烟叶与其他时间发酵烟叶的差异代谢物中均被富集到,且在Nt2016呈现高丰度的差异代谢物,如丙三醇-1-豆蔻酸、肉毒碱、1-棕榈酰-2-氢氧根-丙三基-3-磷酸乙醇胺、a-羟基十四酸等也确实参与脂肪酸降解途径。已有研究表明,脂肪酸降解物与多个烟草评析指标,如香气、烟气浓度、杂气、刺激性、吃味、劲头、甜度等均存在显著或极显著相关水平[31]。结合陈化不同年份烟叶的内在质量评吸结果,香气质和香气量在Nt2016烟叶内在品质达到最佳,因此可以推断,上部烟叶陈化发酵4年时期,烟叶中以假单胞菌属和泛菌属为代表的微生物活性达到峰值,通过脂肪酸降解途径相关的代谢物积累和作用,在提高烟叶化学成分协调性的基础上显著增加了烟叶香气质和香气量,相对于发酵1年和7年来说应为发酵最佳时期。前人研究中曾采用质量评定和化学分析方法探究烟叶陈化发酵过程中生化特性的变化规律,表明不同等级烟叶的理论陈化标准时间有所不同。如余校芳认为上桔二最佳贮存期为12~20个月,上桔三为15~23个月[32];郭俊成等认为中三中四烟叶陈化时间两年为宜,上二烟需要3年时间品质才能达到要求[33]。本研究选用的样品即是内含物质较丰富的上部叶片,其相较于中下部叶片结构更紧密,身份更为厚实,其陈化时间理论上应比中下部叶片更长。另外,韩锦峰的研究表明在现有技术条件下烤烟达到最佳吸味品质的最佳自然醇化时间为24~30个月[34]。由于本研究中取样范围较大,主要比较了0年、1年、4年和7年的陈化样本,因此在后续实验中还可采用微生物组和代谢组研究手段在1~4年时间段内进一步研究烟叶陈化发酵机理。
4 结论
本研究发现在烟叶复烤后的不同陈化发酵时间,烟叶中微生物菌群与代谢产物均会发生显著变化。其中,发酵4年的烟叶中微生物种类最多,多样性丰值最大,且发酵4年烟叶中显著高丰度的差异代谢物主要富集在脂肪酸降解途径。结合感官质量评价数据表明,上部叶片陈化发酵4年相对于其他发酵时期烟叶的内在品质最佳。本研究明确了复烤烟叶在陈化发酵不同时间微生物多样性及代谢物质的差异,有利于烟叶发酵过程中功能微生物的筛选,对烟叶发酵过程的机理研究提供一定理论参考,为进一步研究烟叶陈化时期与烟草吸味品质之间的关系提供了一定理论基础。
[1] 单宏英. 陈化烟叶表面有益微生物的分离筛选、鉴定及应用研究[D]. 杨凌:西北农林科技大学,2012.
SHAN Hongying. Studies on screening, identification and application of beneficial microorganisms isolated from aging tobacco surfaces[D]. Northwest A&F University, 2012.
[2] 陈伦旺. 陈化烟叶微生物的分离鉴定及其在烟叶发酵中的应用[D]. 杨凌:西北农林科技大学,2020.
CHEN Lunwang. Isolation and identification of microorganisms in aged tobacco leaves and their application in tobacco leaf fermentation[D]. Northwest A&F University, 2020.
[3] 赵铭钦,刘云,李芳芳,等. 陈化烤烟叶面优势菌的筛选鉴定与其增香效应[J]. 微生物学报,2009, 49(5): 625-631.
ZHAO Mingqin, LIU Yun, LI Fangfang, et al. Identification of dominant and fragrance-enhancing microorganisms of tobacco leaves during ripening[J]. Acta Microbiologica Sinica, 2009, 49(5): 625-631.
[4] 伍雪莹. 陈化烤烟烟叶细菌群落鉴定及应用研究[D]. 广州:华南理工大学,2014.
WU Xueying. Identification and application study of bacterial communities on flue-cured tobacco leaves[D]. South China University of Technology, 2014.
[5] Fakruddin M, Mannan KS, Andrews S. Viable but nonculturable bacteria: food safety and public health perspective[J]. ISRN Microbiology, 2013, 2013:703813.
[6] Tamang JP, Watanabe K, Holzapfel WH. Review: Diversity of microorganisms in global fermented foods and beverages[J]. Frontiers in Microbiology, 2016, 7:377.
[7] Huws SA, Edwards JE, Kim EJ, et al. Specificity and sensitivity of eubacterial primers utilized for molecular profiling of bacteria within complex microbial ecosystems[J]. Journal of Microbiological Methods, 2007, 70(3): 565-569.
[8] Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data[J]. Bioinformatics,2014, 30(15): 2114- 2120.
[9] Kechin A, Boyarskikh U, Kel A, et al. A new tool for accurate cutting of primers from reads of targeted next generation sequencing[J]. Journal of Computational Biology: a Journal of Computational Molecular Cell Biology, 2017, 24(11): 1138-1143.
[10] Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 2010, 26(19): 2460-2461.
[11] Edgar RC, Haas BJ, Clemente JC, et al. UCHIME improves sensitivity and speed of chimera detection[J]. Bioinformatics, 2011, 27(16): 2194-2200.
[12] Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data[J]. Bioinformatics, 2010, 26(1): 139-140.
[13] Lan Y, Wang Q, Cole JR, Rosen GL. Using the RDP classifier to predict taxonomic novelty and reduce the search space for finding novel organisms[J]. PLoS One, 2012, 7(3): e32491.
[14] Douglas GM, Maffei VJ. PICRUSt2 for prediction of metagenome functions[J]. Nature Biotechnology, 2020, 38(6): 685-688.
[15] Parks DH, Tyson GW, Hugenholtz P, et al. STAMP: statistical analysis of taxonomic and functional profiles[J]. Bioinformatics, 2014, 30(21): 3123-3124.
[16] Galperin MY, Kristensen DM, Makarova KS, et al. Microbial genome analysis: the COG approach[J]. Briefings in Bioinformatics, 2019, 20(4): 1063-1070.
[17] Wang J, Zhang T, Shen X, et al. Serum metabolomics for early diagnosis of esophageal squamous cell carcinoma by UHPLC-QTOF/MS[J]. Metabolomics 2016, 12(7): 1-10.
[18] Qi D, Brownridge P, Xia D, et al. A software toolkit and interface for performing stable isotope labeling and top3 quantification using Progenesis LC-MS[J]. Omics : a Journal of Integrative Biology, 2012, 16(9):489-495.
[19] Slade WO, Werth EG, McConnell EW, et al. Quantifying reversible oxidation of protein thiols in photosynthetic organisms[J]. Journal of the American Society for Mass Spectrometry, 2015, 26(4): 631-640.
[20] Kuhl C, Tautenhahn R, Bottcher C, et al. CAMERA: an integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets[J]. Analytical Chemistry, 2012, 84(1):283-289.
[21] Boccard J, Rutledge DN. A consensus orthogonal partial least squares discriminant analysis (OPLS-DA) strategy for multiblock Omics data fusion [J]. Analytica Chimica Acta, 2013, 769:30-39.
[22] Zhao J, Zhang D, Yang Y, et al. Dissecting the effect of continuous cropping of potato on soil bacterial communities as revealed by high-throughput sequencing[J]. PloS One, 2020, 15(5):e0233356.
[23] 龚俊,刘玉配,李媛媛. 烟叶表面微生物类群两种检测方法的比较研究[J]. 华东师范大学学报(自然科学版),2016(3): 92-101+114.
GONG Jun, LIU Yupei, LI Yuanyuan. Comparative analysis of microbial communities on tobacco leaves between clone library and high-throughput sequencing[J]. Journal of East China Normal University (Natural Science), 2016(3):92-101+114.
[24] Zhong W, Zhu C, Shu M, et al. Degradation of nicotine in tobacco waste extract by newly isolatedsp. ZUTSKD[J]. Bioresource Technology, 2010, 101(18):6935-6941.
[25] 张颖,杨悦,韦庆慧,等. 鞘氨醇单胞菌的特性及应用研究进展[J]. 化学与生物工程,2021, 38(3):6-13.
ZHANG Ying, YANG Yue, WEI Qinghui, et al. Research progress in characteristic and application of[J]. Chemistry & Bioenginering, 2021, 38(3):6-13.
[26] 冯颖杰,袁岐山,杨宗灿,等. 一株鞘氨醇单胞菌对复烤后烟叶多酚物质的降解作用[J]. 中国烟草学报, 2019, 25(1):19-24.
FENG Yingjie, YUAN Qishan, YANG Zongcan, et al. Effect ofsp. strain on degradation of polyphenols in redried tobacco leaves[J]. Acta Tabacaria Sinica, 2019, 25(1):19-24.
[27] Zhao Y, Zhong GF, Yang XP, et al. Bioconversion of lutein to form aroma compounds by[J]. Biotechnology Letters, 2015, 37(8):1687-1692.
[28] 韦康,黄天辉,白森,等. 液相色谱-串联质谱法测定烟叶中的玉米赤霉烯酮[J]. 当代化工,2019, 48(7):1623-1625+1629.
WEI Kang, HUANG Tianhui, BAI Sen, et al. Determination of zearalenone in tobacco leaves by liquid chromatography-tandem mass spectrometry[J]. Contemporary Chemical Industry, 2019, 48(7):1623-1625+1629.
[29] 朱宏建,高必达,易图永. 烟叶贮藏期霉变原因及防霉技术研究[J]. 安徽农学通报,2007, 13(15):139-141.
ZHU Hongjian, GAO Bida, YI Tuyong. Study on mildewing causes of tobacco in storage stage and its control technologies [J]. Anhui Agricultural Science Bulletin, 2007, 13(15):139-141.
[30] 吴丽君,石凤学,刘晶,等. 烟草香气成分分析研究进展[J]. 中国农学通报,2014, 30(21):251-257.
WU Lijun, SHI Fengxue, LIU Jing, et al. Tobacco aroma analysis review[J]. Chinese Agricultural Science Bulletin, 2014, 30(21):251-257.
[31] 李艳梅. 烟叶在烘烤过程中脂氧合酶活性与脂肪酸、色素降解对品质的影响[D]. 郑州:河南农业大学;2001.
LI Yanmei. The effect of degradation of faty acid and pigment and quality of tobacco leaf during flu-cured[D]. Henan Agricultural University, 2001.
[32] 余校芳,李明三,肖本炎,等. 烟叶最佳储存期研究[G]. 国家烟草专卖局科技教育司. 烟草科学技术成果汇编第三辑(1993~ 1995). 郑州:河南科学技术出版社,1996.
YU Xiaofang, LI Mingsan, XIAO Benyan, et al. Study on the best storage period of tobacco leaves[G]. Science and Technology Education Department of the State Tobacco Monopoly Administration. Third Series of Tobacco Science and Technology Achievements Collection (1993~1995). Zhenzhou: Henan Science and Technology Press,1996.
[33] 郭俊成,程晓蕾,肖厚荣,等. 皖南烤烟陈化研究[J]. 中国烟草,1996(2): 16-17.
GUO Juncheng, CHENG Xiaolei, XIAO Hourong, et al. Study on aging of flue-cured tobacco in southern Anhui[J]. Chinese Tobacco, 1996(2):16-17.
[34] 韩锦峰,朱大恒,杨素勤,等. 不同陈化时期烤烟几种酶活性及相关化学成分的分析[J]. 中国烟草科学,1999(1):1-2.
HAN Jinfeng, ZHU Daheng, YANG Suqin, et al. Analysis of enzyme activity and relevant chemical components of flue-cured tobacco in different stages of aging [J]. Chinese Tobacco Science, 1999(1):1-2.
Analysis of microbial diversity and metabolome of flue-cured Tobacco with different fermentation time
ZHANG Wenyou1, YAN Lang1, LAI Xianjun1*, DUAN Wangjun2, ZHANG Yizheng3
1 Sichuan Key Laboratory of Featured Crop Research and Utilization, College of Agriculture Science, Xichang University, Liangshan, 615013, China;2 China Tobacco Sichuan Industrial Co.Ltd., Chengdu 610066, China;3 Sichuan Key Laboratory of Molecular Biology and Biotechnology, College of Life Sciences, Sichuan University, Chengdu 610064, China
[] This study aims to understand the relationship between microorganisms and metabolites in tobacco leaves with different fermentation time and to explore the influence of microorganisms on tobacco leaf fermentation quality. [] Using Illumina high-throughput sequencing technology and UPLC-ATOF MS liquid-mass spectrometry technology were used to analyze the microbial diversity and differentially expressed metabolites of flue-cured tobacco leaf samples fermented for 0, 1, 4 and 7 years. [] Four-year-fermented tobacco leaves had the most abundant microbial species, which showed the highest microbial diversity. At the level of genus,andare the dominant genus in tobacco leaves in samples fermented for 4 years. Combined with the OPLS-DA model and database searching, a total of 173 significantly different metabolites were identified by pair-wise comparison in 4 fermentation stages. After KEGG enrichment, it was found that the different metabolites were involved in amino acid metabolism, fatty acid metabolism, and steroid biosynthesis. Among them, the high-abundance differentially expressed metabolites in the samples with 4 years of fermentation were mainly enriched in the fatty acid degradation pathway. [] There were significant differences in microbial diversity and metabolites of flue-cured tobacco leaves with different time of fermentation, in which four-year fermentation is the best duration of fermentation.
flue-cured tobacco; fermentation; microbial diversity; metabolomics; tobacco quality
. Email:laixianj@hotmail.com
张文友,颜朗,赖先军,等. 陈化烤烟叶片不同发酵时间微生物多样性及代谢组分析[J]. 中国烟草学报,2022,28(5).
ZHANG Wenyou, YAN Lang, LAI Xianjun, et al. Analysis of microbial diversity and metabolome of flue-cured Tobacco with different fermentation time[J]. Acta Tabacaria Sinica, 2022,28(5).
10.16472/j.chinatobacco. 2021.143
四川中烟工业有限责任公司项目“宽窄‘润甜香’核心烟叶原料内涵挖掘与配套技术研究(微生物菌肥修复烟草连作障碍与技术示范方向)”(No. yl2020001)
张文友(1969—),本科,副教授,主要研究方向:烟草栽培学,Tel:0834-2580077,Email:892748686@qq.com
赖先军(1986—),Tel:0834-2580077,Email:laixianj@hotmail.com
2021-07-26;
2022-06-30
