高效凝胶渗透色谱法测定聚乙二醇分子量及其分布
2022-11-10程继业周震宇金伟斌陈礼峰姚玲
程继业,周震宇,金伟斌,陈礼峰,姚玲
苏州市药品检验检测研究中心,苏州 215104
聚乙二醇(polyethylene glycol,PEG)又称聚氧乙烯醇,由环氧乙烷与乙二醇在碱性催化剂催化下聚合而成,分子量在200~35000之间,常见种类有PEG 200、PEG 400、PEG 2000、PEG 4000以及PEG 6000等。PEG具有优良的润滑性、保湿性、分散性、生物相容性,在制药以及日用化学等领域应用广泛。低分子量PEG(分子量为200~400)常作为软胶囊剂、注射液、滴眼剂中的溶剂或助溶剂以增加药用成份的溶解度[1];中高分子量PEG(分子量为2000~8000)在片剂、栓剂、经皮制剂中常作为基质、骨架材料,发挥缓控释作用[2]。不同分子量的PEG对临床疗效、制剂的释药性能具有重要影响[3-5]。
目前各国药典收载的PEG类化合物分子量的测定方法均为酸碱滴定法,通过测定分子链末端氢氧根数量,折算平均分子量。该方法操作过程繁琐,使用的试剂对实验人员和环境毒性较大,影响实验结果的因素较多,并且不能得到分子量分布信息[6-8]。高效凝胶渗透色谱(gel permeation chromatography,GPC)利用体积排阻原理可有效分离PEG化合物,且配合特定的色谱应用软件可对色谱行为进行统计分析并测定重均分子量(Mw)、数均分子量(Mn)、标示分子量(Mp)、分布系数等关键信息,近年来在多分散化合物研究中应用广泛[9-13]。
本研究以国内市售的3家企业多批PEG 3350样品为研究对象,采用GPC技术,对关键的色谱条件及软件处理参数进行系统考察并探讨影响测定结果准确度的各种实验因素,以期为《中国药典》(2020版)中收录的相关高分子化合物分子量分布测定的正确实施提供参考。
1 材料
1.1 药品与试剂
硝酸钠(国药集团化学试剂有限公司,批号:20150508,分析纯);ProClinTM(SIGMA-ALDRICH,批号:MKCH7182);PEG对照品(德国PSS公司,批号:pegkit-08,分子量范围 106~11100);PEG 3350样品9批(A企业产品批号:180810387、180810388、180810389;B企业产品批号:35187、35188、35189;C企业产品批号:4453325、4447021、4480104;编号依次为 A1~A3、B1~B3、C1~C3)。
1.2 仪器
e2695-2414高效液相色谱仪、示差折光检测器、Empower 3 GPC软件(美国Waters公司);XS205DU电子天平(瑞士METTLER TOLEDO公司);Milli-Q超纯水仪(美国Millipore公司);0.22μm微孔滤膜(国药集团化学试剂有限公司)。
2 方法与结果
2.1 色谱条件
Agilent PL aquagel-OH 20色谱柱(300mm×7.5mm,8μm);流动相为0.1mol/L硝酸钠溶液(含0.02% ProClinTM);流速为0.5ml/min;柱温为35℃;检测器温度为35℃;进样体积为100μl。
2.2 溶液的制备
流动相:取8.5g硝酸钠,用超纯水溶解,加入0.2ml ProClinTM,用超纯水稀释至1L,混合均匀,用0.22μm微孔滤膜滤过,即得。
对照品溶液:精密称取不同分子量PEG对照品各50mg,分别用流动相完全溶解并稀释至25ml,摇匀。
供试品溶液:精密称取样品0.1g,用流动相溶解并稀释至50ml,摇匀。
2.3 测定方法
分别取对照品溶液和供试品溶液100μl,按“2.1”项下色谱条件进样分析。其中,对照品设定为窄分布标准进样,供试品设定为宽分布样品进样。对照品属性中输入相应PEG的Mp及分布系数,以主峰保留时间(t)为横坐标、以分子量对数(logM)为纵坐标,拟合三阶校正曲线。基于该校正曲线进行回归计算,得到供试品对应色谱峰的分子量信息(Mn、Mw)及分布系数。
2.4 精密度试验
精密吸取对照品溶液(Mp 3450)100μl,连续进样5次,结果PEG 保留时间的RSD为0.23%。
2.5 重复性试验
精密称取0.1gA1样品,按“2.2”项下方法平行制备6份供试品溶液,结果样品的Mw分别为3363、3349、3358、3362、3354、3341,RSD 为 0.25%。
2.6 进样浓度
考察样品称样量在±20%范围内变化时对结果的影响。取适量A1样品,按“2.2”项下方法分别配制浓度为1.6、1.8、2.0、2.2、2.4mg/ml的溶液并进行测定,结果与“2.5”项下结果基本一致。由于本实验以主峰保留时间作为分子量计算的依据,因此进样浓度在小范围内变化对实验结果无影响。
2.7 线性与范围
取PEG对照品(Mp分别为430、1030、2100、3450、5800),按“2.2”项下方法分别制成对照品溶液,按“2.3”项下方法进行测定(色谱图见图1),并建立三阶校正曲线(图2A)。回归方程为LogM=0.00665t3- 0.372t2+6.59t-33.710, 相关系数R2=0.9999,各校正点的残差均小于2.0%(-0.095%~1.127%),表明分子量在430~5800范围内时线性关系良好。
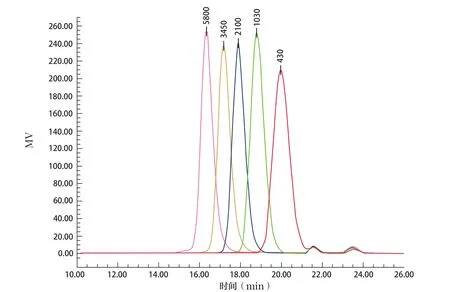
图1 PEG对照品(Mp 430、1030、2100、3450、5800)的色谱图
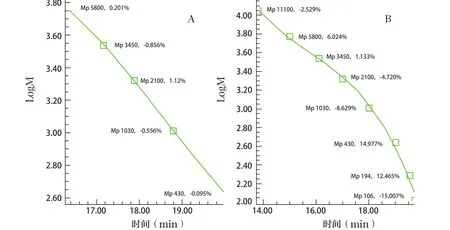
图2 PEG对照品(A:Mp 430~5800;B:Mp 106~11100)的三阶校正曲线
2.8 稳定性试验
取对照品(Mp 3450)溶液和供试品(A1、B1、C1)溶液,室温放置,分别于0、4、8、12、24h进样分析,记录色谱图。结果对照品和供试品的色谱峰形均无改变,峰高均无变化,保留时间的RSD均小于0.53%,表明待测样品溶液在24h内稳定性良好,且色谱系统稳定性也良好。
2.9 准确度试验
与外标法不同,GPC法测定分子量是以主成分峰的保留时间为计算基础,不能用对照品进行回收率试验,因此本研究设计了校正曲线检查,以验证方法的准确性。取Mp为2100、3450和5800的PEG对照品各3份,按“2.3”项下方法分别进行测定,用已建立的校正曲线计算各对照品的Mw,并与Mp比较,计算相对偏差(表1)。3个分子量水平的PEG平均偏差分别为101.41%,99.05%,100.34%,表明方法准确度好。
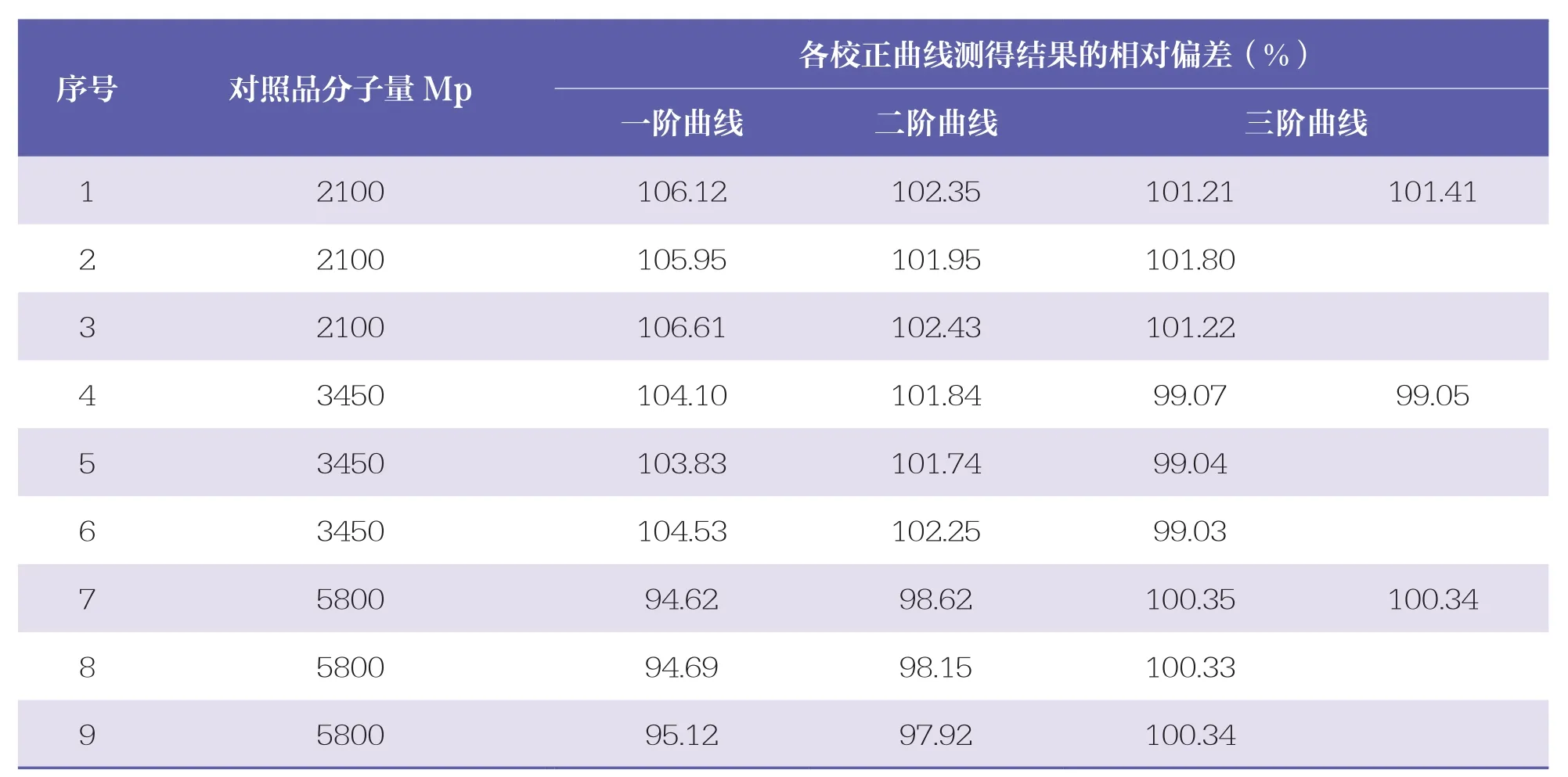
表1 校正曲线的准确度
2.1 0样品测定
取 9 批样品(A1~A3;B1~B3;C1~C3),按“2.2”项下方法制备供试品溶液,并进样测定。结果各批次样品的分布系数均低于1.1,各企业内3批样品间分子量较接近(RSD均低于2%),提示生产工艺稳定。根据产品标准,3家企业的样品均符合规定,但样品分子量差异明显,表现为A企业>B企业>C企业,见表2。
A企业样品均值更接近标示值3350,但分析色谱图发现B、C企业样品色谱峰更对称平滑,而A企业样品色谱峰有小的前沿峰,色谱图见图3。根据体积排阻色谱原理,分子量大的成份应更早洗脱出色谱柱,提示A企业样品中含有一定比例且分子量集中的高分子组份。本研究对GPC系统得到的各批样品累积分布曲线进行比较,依据分子量从高到低,选取10%和90%分位点所对应的分子量,见表2。结果可见,A企业3批样品高分子量区的10%分位点对应分子量大于B、C企业样品,但低分子量区差别较小;A企业样品分子量分布宽度明显大于B、C企业样品,表明B企业和C企业样品的纯度更高。以往方法通过分布系数(Mw/Mn)来评价样品的分布宽度,而本研究表明,分布系数并不能有效评价样品实际分布情况。因此,建议在分子量分布的测定方法中,应根据测得的累积分布曲线增加分布宽度要求以加强对样品多分散属性的控制,例如规定10%~90%分位点对应的分子量宽度。
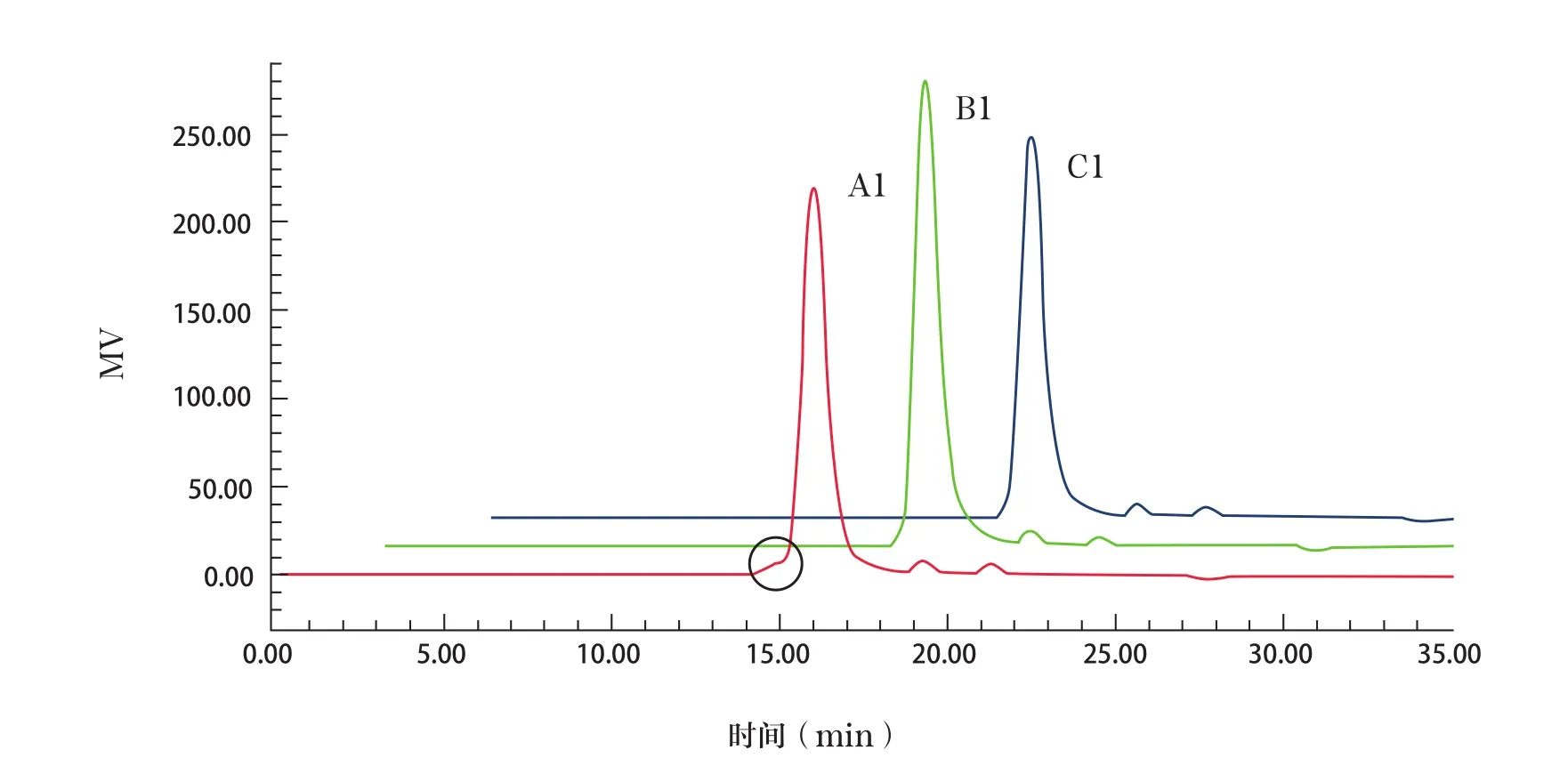
图3 3家企业样品(A1、B1、C1)分子量分布检测色谱图
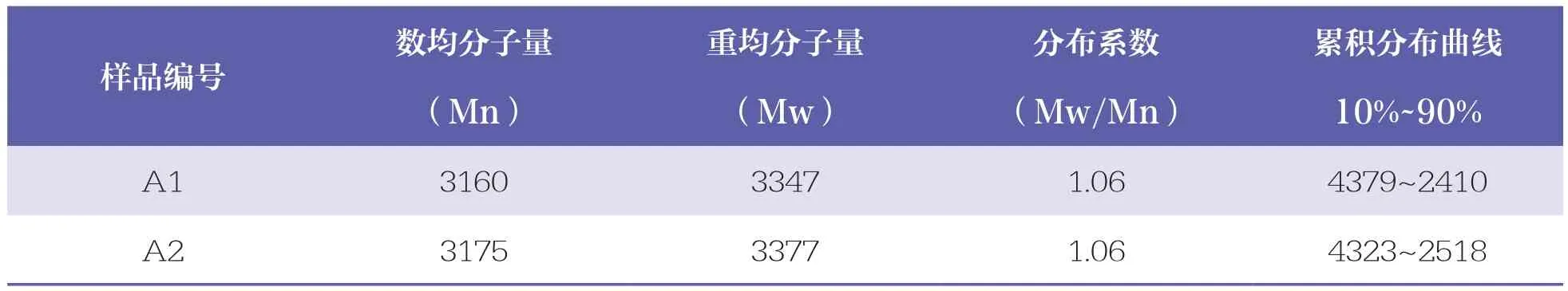
表2 9批PEG 3350的分子量测定结果
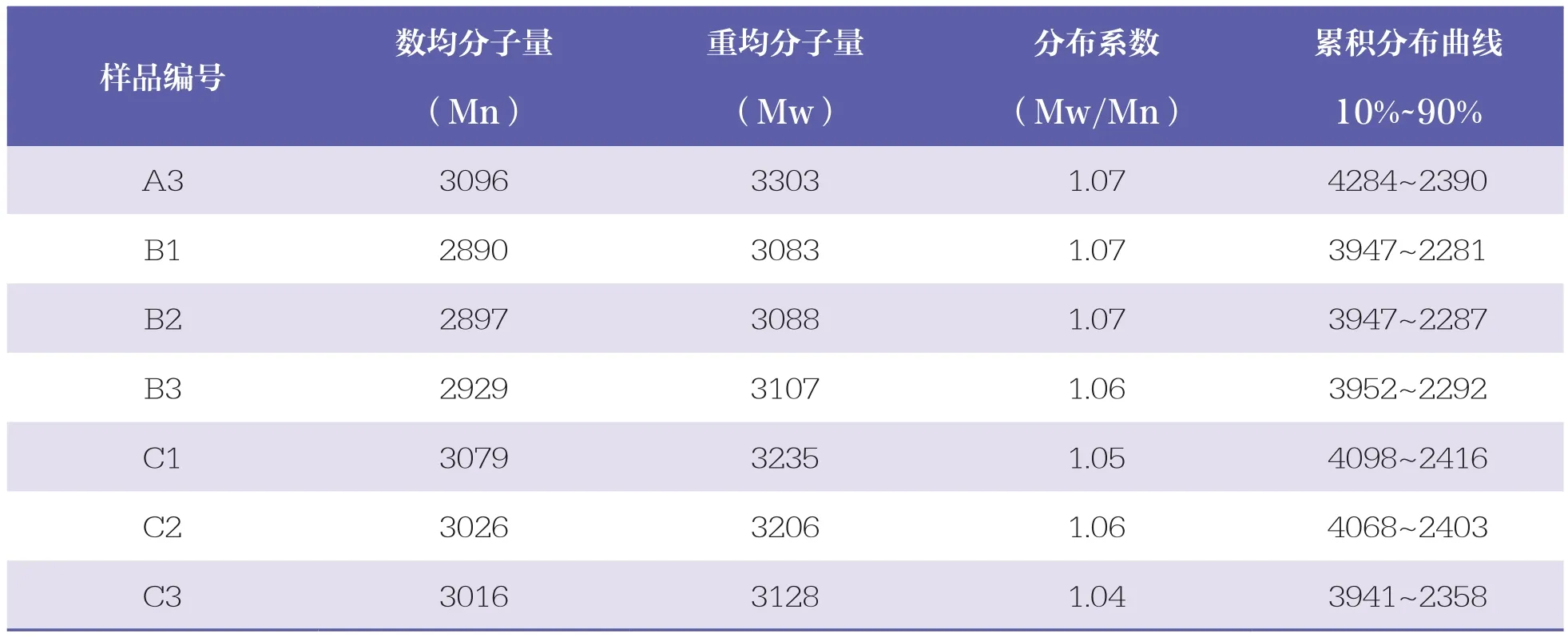
续表
3 讨论
3.1 色谱方法的选择
建立方法时要对分析物的分子量进行初步判断并选择合适的色谱柱[10-11]。本研究对比了Shodex OHpak SB-803 HQ、Agilent PL aquagel-OH 20色谱柱,前者适合分子量为200~40000 Da的PEG类化合物,后者适合100~20000Da。结果表明,Agilent PL aquagel-OH 20色谱柱获得的PEG峰型对称、峰宽较小且对不同分子量PEG的保留区分适当,故选择Agilent PL aquagel-OH 20色谱柱进行实验。
流动相中加入无机盐可维持溶液的离子浓度,获得良好色谱峰形。本研究考察了在流动相中加入0.01~0.5mol/L硝酸钠或者氯化钠时的色谱峰型、柱效及不同PEG分子量的色谱峰分离度情况。结果表明,流动相中加入硝酸钠后基线噪音更小[9];较低浓度(0.01~0.05mol/L)时PEG色谱峰展宽,且不同分子量的主峰分离度下降,而浓度为0.1mol/L时较适宜。本研究所采用的ProClinTM是有机抑菌剂,对人和动物毒性低、对环境友好,与传统分析方法使用的叠氮化钠相比,可有效避免实验过程中对实验人员和环境的危害。
3.2 校正曲线的选择
使用GPC 系统测定化合物分子量时,首先要确定标准曲线类型。本研究比较了一阶~五阶曲线对测定结果准确度的影响,在Mp 2100、3450、5800这3个分子量对照品校正点处,三阶曲线拟合准确度最好(均小于2.0%),一阶和二阶曲线准确度较低,四阶及更高阶曲线由于校正点数少及分子量范围小而不适合本研究,因此选择三阶曲线进行分析。本研究以Mp为430~5800范围内的共5个对照品和Mp为106~11100范围内的共8个对照品分别建立校正曲线,并进行准确度检查。结果表明,图2A中5个点残差均在2.0%以内,图2B中只有Mp 3450处较小,其余各点均大于2.0%,随着分子量减小有明显增大的趋势。该结果表明以Mp为430~5800的5个对照品建立的曲线范围是合适的,且建立方法所用对照品数量和校正曲线的范围应与特定分子量的待测物相适合,利用残差值可对方法测定准确性进行评估。
3.3 对质量标准修订的建议
目前,对于特定分子量的待测样品,如何确定校正曲线的合适范围以及系统适用性试验的指标尚无相关论述[9-13]。《中国药典》(2020版)收载了PEG系列辅料的分子量分布测定方法,但未对方法适用性作出具体要求。对于开展这类工作较少的实验室,方法适用性的缺失不利于评估仪器系统的状态和测试的准确性。基于本研究结果,建议将校正曲线相关参数(如曲线类型、回归系数、残差限度)纳入PEG系列辅料的分子量分布测定方法的系统适用性项下,完善质量标准、提高可执行度。在质量标准中,建议除规定Mw数值和分布系数以外,必要时增加分布宽度要求,例如规定10%~90%分位点处的分布宽度,以加强对产品多分散属性的控制。
4 小结
本研究建立了一种测定分子量在430~5800范围内PEG的GPC方法。该方法结果准确、重现性好,有助于获得Mw、分布系数、累积分布宽度等多分散化合物特征信息,可用于PEG样品的质量控制和比对,在一定程度上为制剂研发中辅料的筛选以及国家标准的提高提供了参考。
