Secondary genomic f indings in the 2020 China Neonatal Genomes Project participants
2022-11-08HuiXiaoJianTaoZhangXinRanDongYuLanLuBingBingWuHuiJunWangZhengYanZhaoLinaYngWenHaoZhou
Hui Xiao · Jian-Tao Zhang · Xin-Ran Dong · Yu-Lan Lu · Bing-Bing Wu · Hui-Jun Wang · Zheng-Yan Zhao ·Lin aYng , · Wen-Hao Zhou,
Keywords Genetics secondary f indings · Neonate · Next generation sequencing
tInroduction
The declining cost of next generation sequencing (NGS),coupled with advanced development of bioinformatics, is creating opportunities to adopt gene sequencing in multiple medical situations, particularly for the molecular diagnosis of rare congenital disease, preconception or prenatal screening, population screening for disease risk, and the individual treatment in cancer. During NGS data interpretation, there is a potential for recognition and reporting of secondary f indings (SFs), which may be of great medical utility to clinicians and patients.
According to the guidelines published by the American College of Genetics and Genomics (ACMG) [ 1, 2], SFs refer to variants with known or possible pathogenicity located in genes that are not directly related to the primary medical conditions. Evidence-based guidelines recommended by ACMG and Association for Molecular Pathology (AMP) in 2015 [ 3] standardized the clinical interpretations, and highlighted that early awareness of SFs may provide clues for disease prevention and intervention at an early stage.
We have previously carried out the China Neonatal Genomes Project (CNGP) [ 4], which is a large observational study aiming to build a Chinese neonatal genome database and to establish a genetic testing workf low for neonatal genetic diseases. Here we report the frequency of secondary f indings obtained from clinical exome sequencing data of 2,020 CNGP patients across the revised list of 59 actionable genes based on the ACMG recommendations for reporting secondary f indings v2.0 (ACMG SF v2.0) [ 2]. This work provided a general analysis of SFs incidence in the Chinese neonatal population and could be used as a reference for early disease intervention.
Methods
Cohort description
This study enrolled cases participating in the CNGP project between June 1, 2019 and December 31, 2020. The CNGP was approved by the Ethics Committee of the Children’s Hospital of Fudan University (CHFudanU_NNICU11) [ 4].
The recruited newborns of CNGP project primarily from Level 3 and 4 Neonatal Intensive Care Unit (NICU) with postnatal age less than 28 days, and their parents were both-Chinese. The CNGP project included patients with one of the following abnormalities: (1) craniofacial deformities;(2) central nervous system anomalies; (3) cardiovascular abnormalities; (4) evidence of metabolic disease; (5) digestive system anomalies; (6) respiratory system anomalies;(7) skeletal abnormality; (8) urinary or reproductive system abnormalities; (9) infection and immune involvement; (10)hematologic abnormalities. These inclusion criteria have been described in detail in our previous published article[ 5]. Newborns with parents that could not make consent decisions, or with parents who rejected genetic data for subsequent research analysis were excluded from this study.All samples used in this study were collected with signed informed consent by patients' parents or legal guardians. A total of 2,020 cases were screened for variants from a list of 59 genes recommended by the ACMG for return of secondary f indings.
Clinical exome sequencing
Brief ly, genomic DNA samples were extracted from whole blood using the QIAamp DNA Blood Mini Kit (Qiagen,Hilden, Germany) following the manufacturer's protocol.Clinical exome sequencing (CES) was generated using the Agilent ClearSeq Inherited Disease panel kit (including 2742 genes). Sequencing was performed on an Illumina HiSeq 2000/2500 platform (Illumina, San Diego, CA), yielding at least 5 Gb of sequencing data per sample. Low-quality reads(reads in which unknown bases are more than 10%, or reads in which more than 50% bases are of sequencing quality lower than 5) were removed from raw fastq data to generate clean reads. Clean reads were then aligned to the reference human genome (UCSC hg19) by the Burrows-Wheeler Aligner (BWA; v.0.5.9-r16), sorted by SAMtools (v.1.8),and removed duplicates by Picard (v.2.20.1). The average on-target sequencing depth was at least 100 × (4.5 Gb of sequencing data). Variants were obtained using GATK [ 6]and subsequently were annotated by ANNOVAR [ 7], VEP[ 8] and Human Gene Mutation Database (HGMD, professional version). Missense variants were assessed with in silico prediction programs SIFT [ 9], PolyPhen-2 [ 10] and MutationTaster [ 11]. Details of the CNGP clinical sequencing pipeline were described in published works [ 12, 13].
Workf low and selection of secondary f indings
In this study, the secondary f indings were def ined as a pathogenic (P) or likely pathogenic (LP) variant in a gene on the consensus list with an associated disease phenotype unrelated to any participant indication. Therefore, variants of the 59 genes recommended by ACMG SF v2.0 were extracted automatically. ForATP7BandMUTYHpresenting genetic model as autosomal recessive inheritance, we would keep only the homozygous or compound heterozygous variants.
Then, the pathogenicity of each variant was reviewed manually and was evaluated according to the 2015 ACMG/AMP Standards and Guidelines by two experts specialized in data analysis and annotation. Corresponding supporting evidence was provided for each variant to assess its pathogenicity. Those variants classif ied as P or LP were selected as returnable secondary f indings. Carrier status for conditions with autosomal recessive inheritance and variants with unknown signif icance (VUS) lacking suffi cient evidence of pathogenicity were not included in this analysis. The detailed classif ication of each pathogenic or likely pathogenic variant is described in Supplementary Table S1.
Results
Population characteristics
A total of 2,020 neonates were enrolled in this study,including 1164 (57.6%) males and 856 (42.4%) females.The median birth weight was 2980 g (interquartile range[IQR], 2064, 3400), and the median gestational age was 37 weeks (interquartile range [IQR], 34, 39). The distribution of gestational age is shown in Supplementary Fig. S1.The top three chief complaints of enrolled neonates were jaundice (575/2020, 28.5%), preterm (520/2020, 25.7%) and respiratory distress (327/2020, 16.2%). During hospitalization, the most aff ected organ systems of these neonates were respiratory system (880/2020, 43.6%), cardiovascular system(567/2020, 28.1%), and digestive system (472/2020, 23.4%).
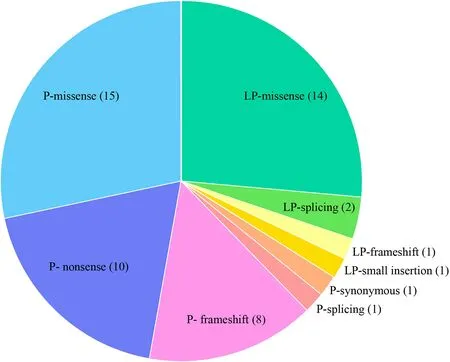
Fig. 1 Distribution of variant types for 53 identif ied variants. P pathogenic, LP likely pathogenic
Secondary f inding results
We identif ied 53 unique variants that accounted for 23 of the ACMG SF reportable genes in 61 individuals, resulting in an overall SF rate of 3.02%. The identif ied variants included 35 pathogenic variants and 18 likely pathogenic variants (Fig. 1). The types of detected unique variants included 29 missense, 10 nonsense, 9 frameshift, 3 splicing, 1 small insertion, and 1 synonymous (Fig. 1). Missense variants accounted for the majority part of P and LP variants, all nonsense variants and the majority of frameshift variants were reported as pathogenic variants.All the P and LP variants determined to be SFs within this cohort arelisted in Supplementary Table S1.
Sixteen genes were detected in more than one patient,including 14 cases inLDLR, each 4 cases in three genes(BRAC1,MYBPC3andMYH7), each 3 cases in four genes(BRCA2,MSH6, FBN1 andTGFBR2) and each 2 cases in eight genes (OTC,PKP2,PMS2,RET,SMAD4,TNNI3,KCNH2andSCN5A) (Fig. 2). In terms of the detected variants, six unique variants in f ive genes (LDLR,TGFBR2,MYH7,FBN1, andBRCA1) were reported more than once(Table 1).
We classif ied the detected genes into four categories by related diseases (Fig. 3). Eleven genes (FBN1,KCNH2,KCNQ1,MYBPC3,MYH7,PKP2,PRKAG2,SCN5A,TGFBR2,TNNI3andTNNT2) related with cardiovascular diseases were detected in 24 patients (1.24%), nine genes(BRCA1,BRCA2,MSH6,PMS2,PTEN,RET,SDHC,SMAD4andSTK11) related with cancer susceptibility and tumor were detected in 19 patients (0.94%),LDLRclassif ied as a gene related to cholesterol and lipid disorders was reported in 14 patients (0.69%), and metabolic disordersrelated genes (GLAandOTC) was reported in 14 patients(0.15%). Obviously, patients identif ied with variants in genes in cardiovascular-related group and cancer-related group occupied the majority of SF reports (44/61).
Impact on medical management and follow-up strategy
The SF results aff ected the medical management and followup strategy in 49 of 61 (80.3%) patients. In terms of genes related to metabolic diseases, one patient withGLAvariant tested plasma α-Gal A activity, and two withOTCvariant completed neurological examination and blood ammonia test instantly for further diagnosis. Twenty-f ive patients with cardiovascular-associated variants and the one withGLAvariant took the echocardiography and were recommended routine echocardiographic follow-up out of concern for changes in the myocardium and cardiac structure. Fourteen patients withLDLRvariants completed the assessment of family member variation; their follow-up strategies and statin treatment plans were formulated according to total cholesterol level and family history of cardiovascular disease.Among cancer-associated genes, tumors caused byPTEN,RET, SDHC,SMAD4, andSTK11may occur in childhood.Seven patients carrying variants in these genes were referred to the oncology department for specialized evaluation and follow-up, and their parents were recommended to improve parental validation.
Discussion
A large number of secondary f indings based on diff erent candidate gene lists and diff erent research races have been reported during the past few years. Previous reports on secondary f indings focused mainly on patients with highly suspected genetic disorders and a large age span. The previously reported SF rates ranged widely because they mainly focused on individuals ascertained on specif ic phenotypes[ 14]. In our study the recruited newborns of CNGP project primarily from Levels 3 and 4 NICU with relative minimal clinical presentation selection. There may be some diff erences in the carrying of genetic variation in neonates compared with children and adults. We identif ied 61 individuals carrying pathogenic or likely pathogenic variants among the 2020 CES datasets, resulting in an SF rate of 3.02%.
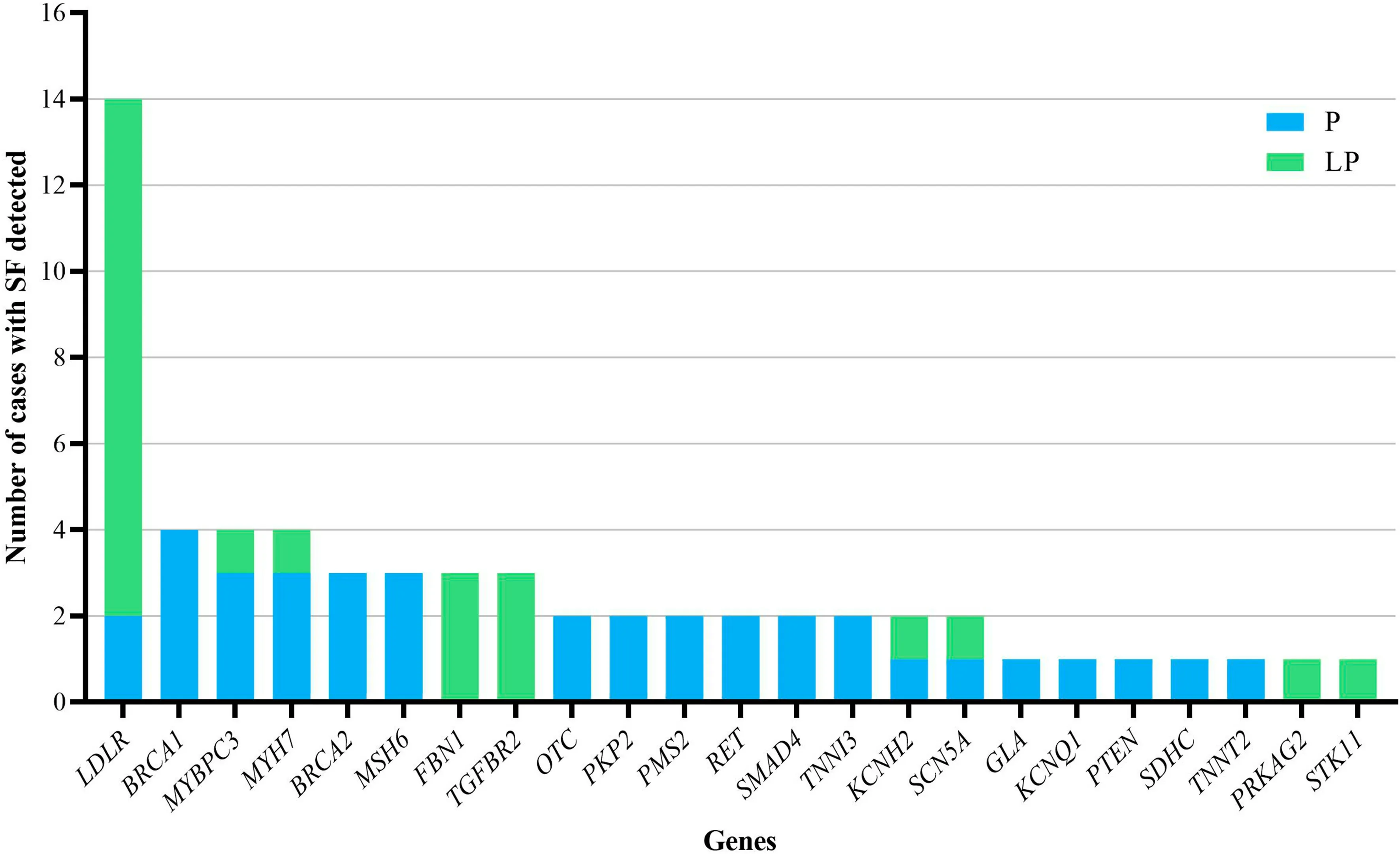
Fig. 2 The number of cases with secondary f indings (SF) detected for each gene. P pathogenic, LP likely pathogenic
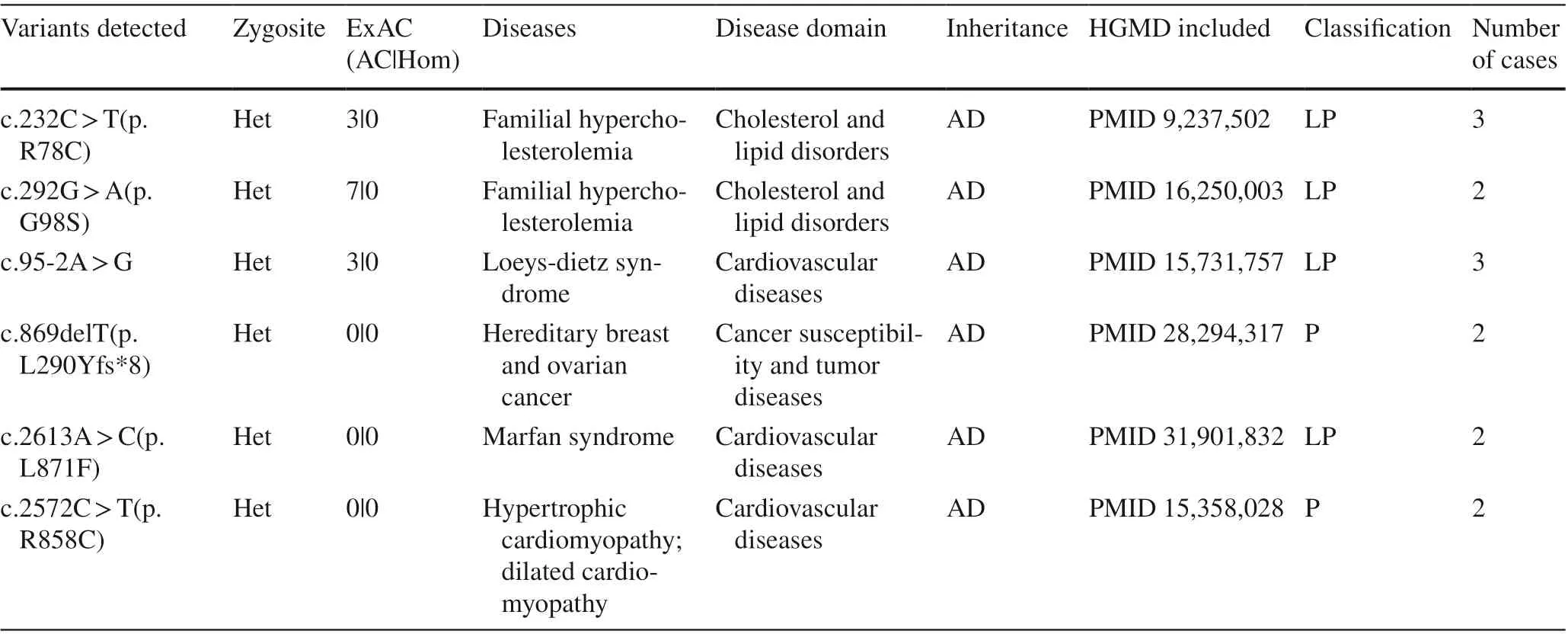
Table 1 Secondary f indings variants identif ied in ≥ 2 participants
According to the characteristics of our population, hospitalized neonates were sent for genetic test mainly because of jaundice, premature delivery, and neonatal respiratory distress. Neonates often present with atypical clinical phenotypes. Although the secondary f indings cannot exactly explain the current phenotypes of the patients, they may help to improve clinical management and follow-up strategies. We reported 35 pathogenic variants and 18 likely pathogenic variants as SF in this study. All nonsense variants and almost all frameshift variants were pathogenic variants. Frameshift and nonsense variants have a greater impact on protein function, and therefore added higher pathogenic evidence when manually evaluated the pathogenicity.
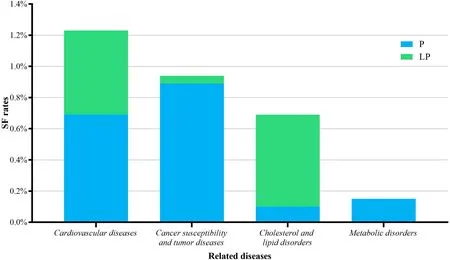
Fig. 3 Secondary f indings (SF) rates classif ied by related disease
The cancer-associated genes that we detected usually cause disease in adulthood. Previous standards for predictive genetic testing recognized a distinction between providing results to adults and to children, which suggested that predictive testing for adult-onset diseases may not be off ered to children [ 15]. However, according to ACMG recommendations [ 1], results from genetic testing of a child may have implications for the parents and other family members. It was recommended that seeking and reporting secondary f indings without restricting the age of the person being sequenced. The detection of pathogenic variants in cancer-associated genes of neonates may present limited diagnosis value or changing clinical management in the current neonatal period, but the results provide a reference for cancer susceptibility study, pedigree analysis, and adult cancer intervention to parents.
LDLRwas the most frequently detected gene in our study,classif ied as a gene related to cholesterol and lipid disorders and causing familial hypercholesterolemia (FH). The high frequent variants in our study were c.232C > T and c.292G > A, which were recorded in HGMD but not reported in children or adolescent patients with heterozygous variant.It is diffi cult to diagnose FH in childhood because children do not have typical clinical phenotypes as xanthomas and corneal rings except homozygous FH. In addition, it is hard to conf irm their detailed family history of FH or premature coronary artery disease [ 16]. In terms of newborns, their serum lipid level may be aff ected by the mother’s pregnancy complications; and the typical FH phenotype is rarely seen during the neonatal period. Thus, genetic analysis demonstrates clinical utility for assessing the risk of FH in neonatal and parents. Previous studies have shown that FH children with pathogenic variants tend to have higher serum LDL-C levels, which are aff ected by the types of variants. For any LDL-C level, FH variant carriers are at increased risk of cardiovascular disease compared to non-carriers [ 17]. Even a low LDL-C level measured at one specif ic time-point in children with FH should need further monitoring. Therefore,for neonates with SFs reported inLDLR, it is recommended to closely follow their serum cholesterol level and the incidence of cardiovascular disease in family members, which may be benef icial to early diagnosis and statin treatment for reducing the lifelong LDL-C burden and the risk of coronary heart disease [ 18, 19].
SFs of cardiovascular-associated genes are mainly related to cardiomyopathy, aortopathy, and long QT syndrome(LQTS). In our study the most frequent genes in cardiomyopathy wereMYBPC3andMYH7. As the main pathogenic genes of hypertrophic cardiomyopathy (HCM), approximately 70% of HCM-causing variants occur in one of them[ 20]. There was no clear correlation between variant type or location and phenotypic severity or outcome. However, progressive left ventricular hypertrophy were found in pediatricMYBPC3variant carriers with non-obstructive arrhythmia phenotypes, and increased risk of death or transplantation were found in children with HCM diagnosed during infancy [ 21, 22]. Therefore, it is recommended to improve HCM screening for infants with HCM-related SFs, especially in families with a history of cardiomyopathy. Other cardiovascular-associated genes also cause disease with a wide variety of symptoms and prognoses. For example,KCNH2,KCNQ1, andSCN5Aare related to LQTS, of which patients may present with bradycardia and atrioventricular block in early infancy or with no symptoms or normal life expectancy [ 23, 24]. Because the pathogenic variants of cardiovascular-associated genes may cause diseases with unrecognizable manifestations or even poor prognosis such as sudden cardiac death, as well as most of these related diseases also exhibit phenotypic heterogeneity and agerelated penetrance, a long-term cardiac follow-up is necessary [ 24— 26].
Metabolic-associated genes in our study includedGLAandOTC.GLAis the causative gene of Anderson-Fabry disease (AFD), which is a rare X-linked multi-tissues lysosomal storage disorder [ 27]. In classically affected males, it leads to the onset of angiokeratomas, acroparesthesias, hypohidrosis, and a characteristic corneal opacity early in childhood or adolescence; renal insufficiency,cardiac and cerebrovascular disease may appear with age advancing [ 28]. In our study, a c.640-801G > A variant inGLAwas identified in a male newborn. Large newborn screening studies have shown that this variant was highly prevalent in the Taiwanese population [ 28— 30]. Given the unignorable severity, this patient should take α-Gal A activity testing for further diagnosis confirmation and early treatment initiation; and more attention should be paid to cardiac abnormality in his future [ 31, 32].OTCis the causative gene of ornithine transcarboxylase deficiency (OTCD). In affected males, the disease occurs as a neonatal or late-onset hyperammonemic coma, associated with severe liver and brain sequelae or even death [ 33,34]. Heterozygous females are symptomatic in around 20% of cases [ 35, 36]. In this study, we reported two female patients carrying c.118C > T and c.622G > A inOTC,respectively. These two variants were classified as pathogenic in HGMD for fatal late-onset OTCD in boys,while without cases of heterozygous females reported.High clinical heterogeneity in OTCD is possibly related to inter-individual variability of mutant X chromosome inactivation [ 37], the degree of skewness may result in different levels of residual enzyme activity, even females harboring the same variant have variable clinical expression [ 36]. Hence, the severity of the disease is unpredictable in heterozygous females. The SFs of theOTCin these two girls suggested standing monitoring of their nervous syndrome and blood ammonia during their growth.
Therefore, when clinicians receive genetic test report of the hospitalized neonates, they can pay attention to the possible changes in the future according to the disease corresponding to the SF and can conduct early multidisciplinary cooperation and discussion in a long-term follow-up process.Early intervention and monitoring based on secondary f indings may alleviate the problem and may postpone the onset of symptoms of some patients, which presents certain clinical signif icance.
There were two limitations in this study. First, the SF rate may be underestimated because we used clinical exome sequencing as our method; other genetic sequencing methods, such as whole genome sequencing, were able to detect more variants. The samples in our cohort were insuffi cient of family member information, which made it hard to identify and evaluate the de novo variant in detail; second, the enrolled participants were neonates hospitalized in our hospital who were not representative of the entire Chinese neonatal population. As a large proportion of high-risk neonates in our population, the higher possibility of carrying pathogenic variants may have aff ected our results. Parents of normal newborns rarely ask for genetic testing, so there were little genetic sequencing data for normal newborns; and the SF incidence in the normal neonatal population was unavailable. The actual SF rate of the Chinese neonatal population requires further study of random samples.In summary, we found a 3.02% overall frequency of SFs in a cohort of 2,020 CNGP participants. The most common disease category of SFs in our cohort was cardiovascular disease, followed by cancer and tumor susceptibility. and these two groups included the majority of genes on the SF list. Our study about SFs generally demonstrated that SFs are not rare in the Chinese neonatal population. Even if SFs are not f irst-tier targets of the original sequencing, they are still of great practical value in monitoring and intervention of late-onset diseases at an early stage.
Supplementary InformationThe online version contains supplementary material available at https:// doi. org/ 10. 1007/ s12519- 022- 00558-w.
AcknowledgementsWe are grateful for the willingness and cooperation of the patients and their families in this study. The authors also wish to acknowledge various doctors in NICU and Molecular diagnostic center of Children’s Hospital of Fudan University, and the contributing members of the “China Neonatal Genomes Project (CNGP)”.
Author contributionsHX designed the study, collected data, drafted the initial manuscript, reviewed the manuscript and revised the manuscript. JZ, XD, YL, BW and HW designed the data collection instruments, performed the initial analyses, and were involved in writing the manuscript. ZZ, LY and WZ were involved with the study design,supervised data collection, and critically reviewed the manuscript for important intellectual content. All authors approved the f inal manuscript as submitted and agreed to be accountable for all aspects of the work.
FundingShanghai Municipal Science and Technology Major Project(Grant No.20Z11900600) and Clinical Research Plan of Shanghai Hospital Development Center (SHDC2020CR6028-002).
Data availabilityThe datasets analyzed during the current study are available from the corresponding author on reasonable request.
Declarations
Ethical approvalThis study was approved by the Ethics Committee of the Children’s Hospital of Fudan University, and informed consent was obtained from all the study participants.
Conflict of interestAuthor Zheng-Yan Zhao is the Chief Editor forWorld Journal of Pediatrics. The paper was handled by the other Editor and has undergone rigorous peer review process. Author Zheng-Yan Zhao was not involved in the journal's review of, or decisions related to, this manuscript. All other authors declare no competing interests.
杂志排行

World Journal of Pediatrics的其它文章
- Single ventricle: amphibians and human beings
- Clinical diff erences among racially diverse children with celiac disease
- Effi cacy of live attenuated vaccines after two doses of intravenous immunoglobulin for Kawasaki disease
- Associations between body mass index in diff erent childhood age periods and hyperuricemia in young adulthood: the China Health and Nutrition Survey cohort study
- Frequency and safety of COVID-19 vaccination in children with multisystem inf lammatory syndrome: a telephonic interview-based analysis
- Music therapy and pediatric palliative care: songwriting with children in the end-of-life