稀土氧化物Gd2 O3、Nd2 O3、Sm2 O3和Dy2 O3在KCl-LiCl-Li2 O熔盐中的电解
2022-11-07朱铁建佘长锋
季 男,彭 浩,蒋 锋,2,黄 卫,2,*,朱铁建,佘长锋,龚 昱,2
1.中国科学院 上海应用物理研究所,上海 201800;2.中国科学院大学,北京 100049
随着世界范围内核能产业的发展,近年来乏燃料的处理问题已经成为影响核电可持续发展的关键因素。我国核电发展相对于其他发达国家起步较晚,导致我国核燃料循环技术还较为落后,所以乏燃料后处理就成为我国核电发展中面临的重要问题之一。目前为止对乏燃料的管理主要有3种燃料循环的方式:(1)一次性通过方式;(2)后处理热堆循环;(3)先进核燃料循环[1]。前者完全不处理,后两者则可以通过对乏燃料的处理来实现核燃料的可持续性。常规的燃料处理有两种方式,即水法和干法处理技术。相对采用水溶液和有机溶剂的水法处理技术,基于高温冶金的干法后处理技术(pyroprocessing technology)的优势在于:耐高温和耐辐照;工艺操作流程较易;低临界的风险、放射性废物大大减少和避免了核扩散等。其中,熔盐电解法是目前发展最为广泛的干法技术,它已经成为乏燃料后处理研究中最常见的分离技术方法。然而,熔盐电解法也存在一些尚未解决的问题:(1)所产生的气体对环境存在一定的污染;(2)原料一般为金属的卤化物或一些易于溶解在熔盐中的金属化合物,这些原料获得途径困难且价格昂贵;(3)目前的处理对象主要为金属型乏燃料,而对绝大部分的氧化物乏燃料尚无有效方法[2]。
2000年剑桥大学的Zheng等在CaCl2熔盐中成功实现了直接将TiO2电解还原为金属Ti,这种电解方法被称为FFC剑桥法(FFC-Cambridge)[3-4]。FFC剑 桥 工 艺 以CaCl2作 为 电解液,石墨为阳极,在电解温度和电压分别低于金属熔点和熔盐分解电压条件下电解,最终金属氧化物被还原成金属或合金,而O2-进入熔盐进而迁移至阳极放电,生成氧气和碳氧化合物。这种工艺方法将金属氧化物在熔盐中直接电解还原为金属单质,并且阳极只产生氧气或者气态的碳氧化合物,可以很好解决上述问题。目前FFC剑桥工艺法研究对象所选用的电解液,CaCl2熔盐体系最为普遍,也有选择CaCl2-CaO[5]、CaCl2-NaCl等[6]体系的。但该类熔盐存在熔点和电解温度过高的问题,过高的使用温度不仅提高了能源和材料成本,且会使得阴极更加紧密,阻碍了氧化物阴极中氧离子的迁移。现在对铀的氧化物电化学还原的研究,选择的电解液大多数为低熔点的LiCl-Li2O,而不是CaCl2-CaO。因此,选择具有较低熔点的LiCl体系熔盐,如LiCl-KCl和LiCl-KCl-CaCl2等,同样具有良好的O2-溶解度。Hur等[7]在793 K下的LiCl-KCl熔盐中通过添加Li2O成功将UO2还原为金属铀,还原率高达98%。在LiCl-KCl-Li2O电解液中,阴极(氧化物,MxOy)可能发生反应(式(1)—(5))[8-9]。
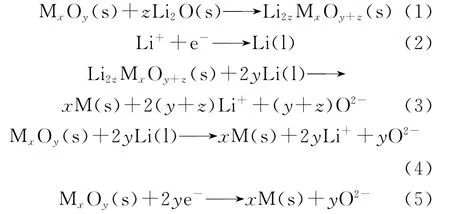
阳极(石墨电极)可能发生的反应如下(式(6)—(8))[10]。

近年来,人们对氧化物乏燃料的电化学还原进行了广泛的研究。氧化物乏燃料中不仅含有需要回收的锕系氧化物,还有待分离的裂变产物,如镧系等氧化物,包括钇、镧、铈、镨、钕、钷、钐、铕、钆、铽十多种稀土元素[10-13]。在电解还原过程中,需要将锕系氧化物还原为金属,而镧系裂变产物氧化物的还原行为会直接影响电解还原产物的组成,进而也影响后续的电解精炼过程和精炼产物的去污效果。因此,需要关注镧系裂变产物氧化物在电解还原过程中的电化学行为。目前在FFC工艺中以低熔点的LiCl-KCl为电解液,将稀土氧化物电解还原为金属单质的方法尚没有开展较多的研究。
本工作拟首先研究LiCl-KCl熔盐中Li2O的电化学行为,并选取Gd2O3、Nd2O3、Sm2O3、Dy2O3作为阴极,验证它们在LiCl-KCl-Li2O熔盐中电解还原的可能性。结合X射线衍射分析(XRD)表征手段,分析氧化物电极和电解产物的物相组成及电解过程的反应原理,并利用氧分析仪测试电解产物的氧含量,计算得到相应的电解还原率。最后利用氧化物电解还原的理论模型,计算稀土氧化物电极的最优孔隙率和最短电解时间,并与实验结果进行比较和分析。
1 实验部分
1.1 试剂和仪器
稀土氧化物Gd2O3、Nd2O3、Sm2O3、Dy2O3,纯度≥99.9%;粘结剂聚乙二醇,化学纯;LiCl,纯度≥97.0%;KCl,纯度≥99.5%;Li2O,纯度≥99.99%。以上药品均采购于国药集团化学试剂有限公司。Mo丝,直径d=1 mm,纯度≥99.99%,哈尔滨碘钨丝厂;石墨棒,d=5.0 mm,纯度≥99.99%,烟台美尔森石墨有限公司。
BJ-15压片机,天津博君科技有限公司;Autolab PGSTAT 302N电化学工作站,瑞士Metrohm有限公司;IT6302直流稳压电源,艾德克斯;Pore-Master33GT压汞仪,Quantachrome;X′Pert Pro MPD XRD,荷兰帕纳科公司;O836氧成分分析仪,美国LECO公司。
1.2 熔盐的制备和净化
实验所用熔盐由33.6 g的LiCl和46.4 g的KCl混合而成。首先,将80 g低共熔点LiCl-KCl混合物置于马弗炉中升温至773 K,保温2 h,再将温度降至473 K保温72 h。实验前,将干燥后的LiCl-KCl熔盐放于刚玉坩埚中,在773 K下,通过恒电位-2.0 V(vs Ag/AgCl)预电解3 h,除去熔盐中的杂质离子和残留水分。
1.3 电化学测试
电化学行为测量时采用三电极测量体系,由参比电极、工作电极、辅助电极(对电极)组成。参比电极为Ag/AgCl(w=1%,摩尔分数为0.39%),内参比盐是LiCl-KCl。工作电极为惰性Mo电极。辅助电极为石墨棒。实验开始前,采用SiC砂纸对惰性Mo电极进行打磨抛光,然后置于稀盐酸中浸泡,再用丙酮进行超声清洗,烘干备用。而石墨棒则需经过稀盐酸煮沸和去离子水清洗处理,并烘干后备用。
电解过程中采用两电极体系,由工作电极和辅助电极(对电极)组成。辅助电极依然是石墨棒,工作电极则为稀土氧化物电极。工作电极的制作方法为:分别称取1.2 g的稀土氧化物(Gd2O3、Nd2O3、Sm2O3、Dy2O3)置于烧杯中,添加适量的聚乙二醇和去离子水,混合均匀。烘干后将干料放入玛瑙研钵中充分研磨后置于模具中,并于压片机15 MPa下保压5 min,脱模取出生坯。将生坯置于马弗炉中,高温烧结7 h。整个升温过程如下:(1)将温度缓慢升至573 K保温3 h,目的是将添加剂聚乙二醇分解完全;(2)从573 K缓慢升温至973 K保温7 h;(3)从973 K缓慢降温至室温。将烧结好的胚体用钼丝捆绑固定制成阴极待用,电极的直径d=15 mm,厚度l=2 mm。
使用Autolab PGSTAT 302N电化学工作站和Nova 2.1软件进行电化学行为测试,包括循环伏安法和计时电位法。使用直流稳压电源进行稀土氧化物电解,采用恒槽电压法,电解示意图示于图1。所有实验均在手套箱内进行(O2和H2O体积分数均小于1×10-6)。
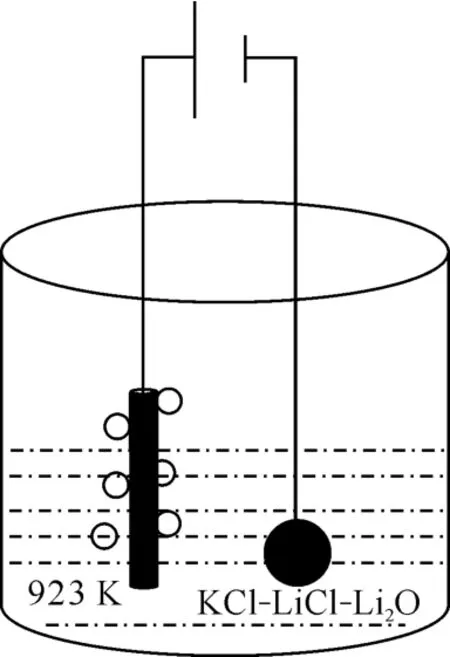
图1 电解示意图Fig.1 Schematic diagram of electrolysis
1.4 表征
烧结的氧化物片体采用压汞仪分析孔隙率,采用XRD,在40 k V和40 m A下使用Cu Kα辐射分析物相组成。电解产物用蒸馏水洗涤,真空干燥后同样使用XRD分析物相组成。采用氧成分分析仪测定电解前后的阴极氧含量。
2 结果与讨论
2.1 KCl-LiCl-Li2 O熔盐的循环伏安和计时电位曲线
923 K、KCl-LiCl熔盐体系中添加Li2O(w=1%)前后在惰性Mo电极上的循环伏安曲线(三电极体系:工作电极、参比电极、对电极)示于图2(a)。其中,虚线是未添加Li2O的空白熔盐,由图2(a)可知:曲线存在一对氧化还原峰A/A′,峰A对应熔盐中Li(Ⅰ)的还原(Li++e-→Li),还原电位-2.40 V(vs Ag/AgCl);峰A′对应金属Li的氧化(Li→Li++e-),氧化电位-2.03 V(vs Ag/AgCl);实线是添加了1%Li2O的KCl-LiCl熔盐,除了氧化还原峰A/A′外,又出现了峰B/B′和C′。而Li2O在熔盐中是以溶解离子的形态存在,在KCl-LiCl熔盐体系中可以提供Li+和O2-[14],所以,氧化峰C′峰电位为-0.55 V(vs Ag/AgCl),发生的反应为2O2-→O2+4e-。因为阴极Mo与生成的O2反应形成MoO2,所以在-1.71 V(vs Ag/AgCl)处的还原峰B和-1.49 V(vs Ag/AgCl)处的氧化峰B′可能对应着Mo电极的氧化还原,所发生的反应分别如式(9)和(10)。
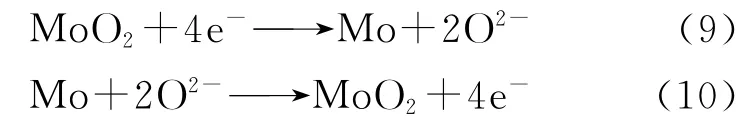
Gonzalez等[15]提到在923 K下,LiCl熔盐体系中以W作为阴极添加Li2O,在循环伏安曲线中阴极和阳极分别发生的电化学反应为WO2+4e-→W+2O2-和W+2O2-→WO2+4e-。这与本实验的结果相似。
相应熔盐体系的计时电位曲线(三电极体系)示于图2(b)。由图2(b)可以观察到两个明显的电位平台A和B,它们分别对应Li(Ⅰ)还原为Li和MoO2还原为Mo的过程。平台A和B的沉积电位分别为-2.24 V和-1.67 V(vs Ag/AgCl),与对应的循环伏安还原峰电位相吻合。
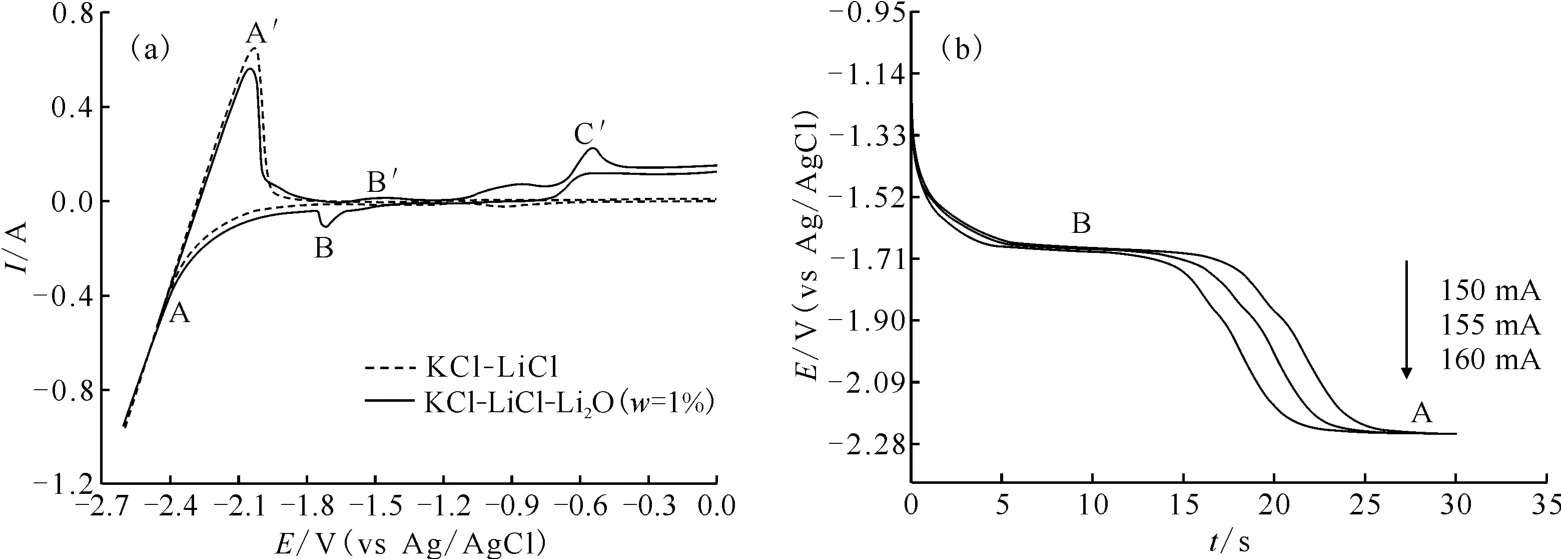
图2 LiCl-KCl熔盐中添加Li2 O(w=1%)前后的循环伏安曲线(a)以及LiCl-KCl熔盐中添加Li2 O(1%)的计时电位曲线(b)Fig.2 Cyclic voltammograms attained in LiCl-KCl molten salt before and after adding Li2 O(w=1%)(a),and chronopotentiograms obtained in LiCl-KCl molten salt added Li2 O(w=1%)(b)
对阳极O2-的扩散过程展开进一步研究。图3(a)所示为923 K下在LiCl-KCl-Li2O熔盐中0.0—-1.0 V的循环伏安曲线,发现C′所对应的反应是不可逆的。由于图3(a)的循环伏安曲线具有比较大的背景电流,所以直接使用循环伏安法计算O2-扩散系数D会带来比较大的误差,因此对循环伏安曲线进行卷积处理。循环伏安数据的半积分结合式(11)可以获得卷积伏安法曲线[16-18]。
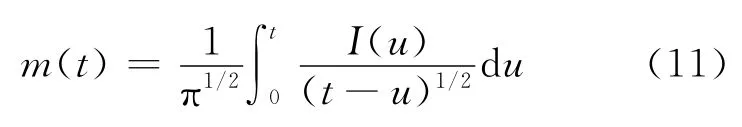
显然根据卷积的定义[19],电流的半积分值,就是电流I(t)与时间函数(πt)-1/2的卷积,所以,可以将m(t)定义为:

式中:*为卷积运算符号;i(t)为循环伏安的电流;m(t)为半积分电流。通过O2-氧化过程中的循环伏安曲线(图3(a))可以获得卷积伏安曲线,示 于图3(b)。据卷积分理论可知[16],m(t)达到极限值m*时,m*可以表示为:

图3 LiCl-KCl-Li2 O熔盐的循环伏安曲线(a)和循环伏安曲线的卷积曲线(b)Fig.3 Cyclic voltammograms attained in LiCl-KCl-Li2 O molten salt(a)and convolution curve of cyclic voltammetry curve(b)
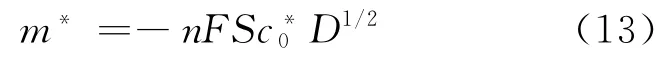
其中:n,转移电子数;F,法拉第常数,96 500 C/mol;S,电极面积,cm2;D,扩散系数,cm2/s;c*0,O的原始浓度,mol/cm3。
通过图3(b)可以获得极限电流m*,并结合式(13)求得扩散系数D。经计算O2-在LiCl-KCl中的扩散系数为0.5×10-5cm2/s。Sakamura[20]提到在923 K下,LiCl-KCl熔盐中O2-的扩散系数值要小于O2-在LiCl熔盐体系中的扩散系数(4×10-5cm2/s),这与本实验中通过卷积伏安法求得的结果一致。
2.2 Gd2 O3、Nd2 O3、Sm2 O3、Dy2 O3的电解
采用恒电压法、两电极体系(工作电极、对电极),在923 K的LiCl-KCl-Li2O熔盐中电解稀土氧化物Gd2O3、Nd2O3、Sm2O3、Dy2O3,且电解过程中熔盐处于静态。通过HSC5.0化学软件结合式(14)分 别 计 算 出Gd2O3、Nd2O3、Sm2O3、Dy2O3、Li2O、KCl、LiCl在923 K下的吉布斯自由能(ΔGf)和相应的分解电位(ΔE),结果列入表1。从表1中各物质的分解电位分析,将电解实验的槽电压设置为3.40 V较为合适。
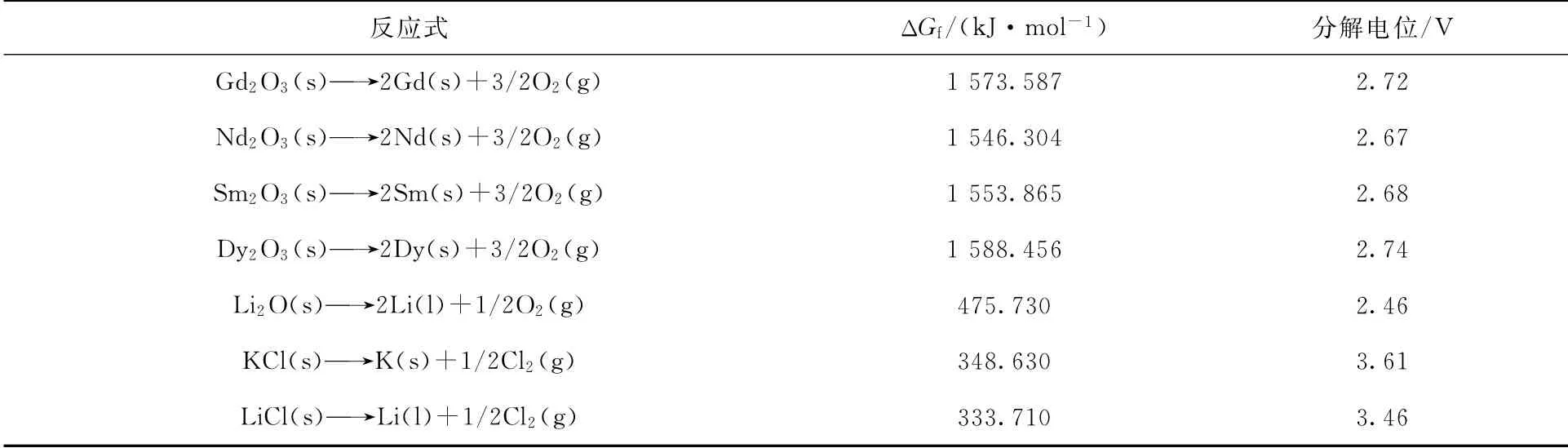
表1 Gd2 O3、Nd2 O3、Sm2 O3、Dy2 O3、Li2 O、KCl和LiCl在923 K下的吉布斯自由能和相应的分解电位Table 1 Gibbs enthalpy of formation and decomposition voltages of chemical compounds Gd2 O3,Nd2 O3,Sm2 O3,Dy2 O3,Li2 O,KCl and LiCl at 923 K

(1)电解前的Gd2O3电极片分析
将Gd2O3片烧结前后的样品进行XRD分析,结果示于图4。由图4可知:Gd2O3烧结前后成分并没有发生改变,说明烧结温度并不影响稀土氧化物的化学形态。孔隙率测试结果表明,烧结后的Gd2O3电极片孔隙率可以达到49.9%。

图4 Gd2 O3烧结前(a)后(b)的产物XRD图谱Fig.4 XRD patterns of Gd2 O3 products before(a)and after(b)sintering
(2)Gd2O3电解
以Gd2O3片为阴极,3.40 V槽电压下电解25 h所得到的时间与电流关系曲线示于图5(a)。由图5(a)可知:电解开始时电流较大,随后降低,这是因为在电解初期,主要反应发生在阴极的表面,离子传输速率较快。当阴极表面的反应完成后,内部的传质速率较慢,电解反应会受物质扩散控制影响,导致反应速率变慢,相应的电流也会降低。在反应初期,O2-浓度足够大,所以电流较大。随着反应的进行O2-的浓度逐渐降低,O2-的传输受到阻碍导致反应速率变慢,也导致相应的电流降低,曲线呈现下降趋势。Xu等[6]在CaCl2-NaCl体系中电解Nb2O3实验中发现添加CaO后的电流值明显大于未添加CaO的,且曲线较陡峭,说明当有足够的O2-时反应进行很快,O2-含量逐渐减少时电流明显下降,最终达到一个平衡状态。这一现象与本实验的结论相似。图5(a)中的插图是电解前后阴极照片,阴极片颜色由白色变成灰黑色,显示有新物质生成,但是电解前后阴极的体积几乎没有变化。电解产物的XRD图谱示于图5(b)。由图5(b)可知:有金属Gd的生成,说明Gd2O3在LiCl-KCl-Li2O熔盐中可以被还原为金属单质。除了金属Gd相和少量的盐(KCl和Li2O)外,阴极中还有中间产物LiGdO2的生成。
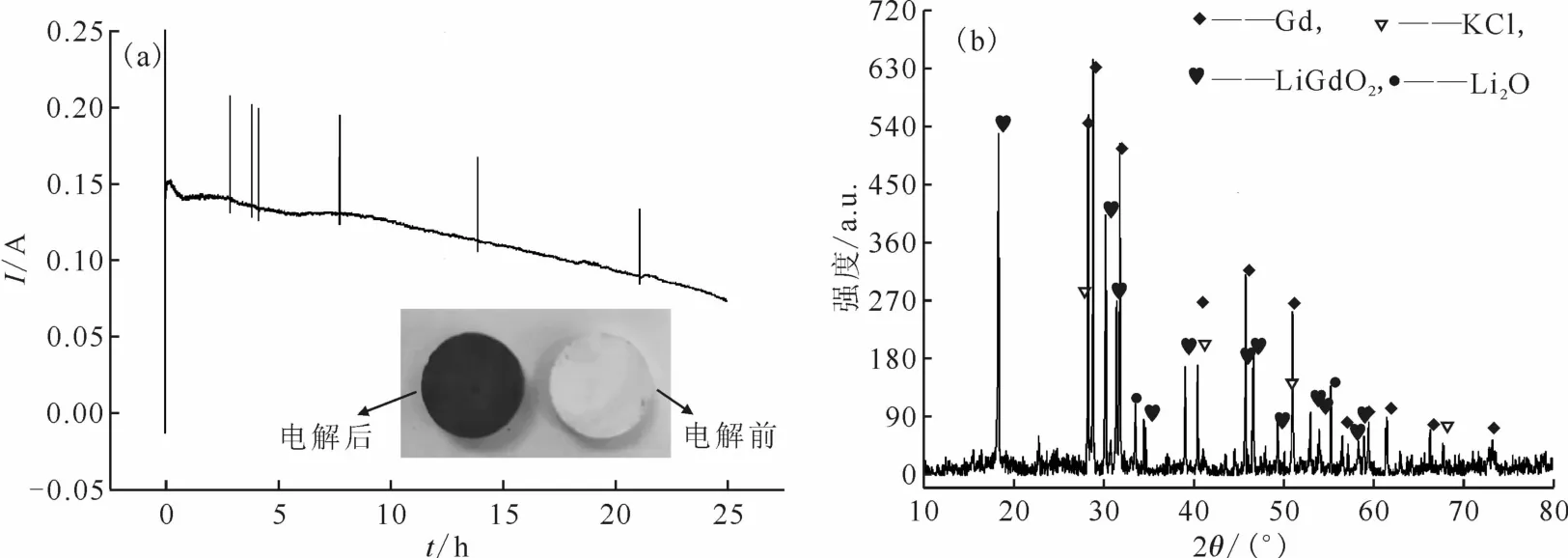
图5 Gd2 O3阴极在KCl-LiCl-Li2 O(w=1%)熔盐中的电流-时间曲线(a)和电解产物的XRD分析图谱(b)Fig.5 Plot of i vs t of Gd2 O3 cathode(a)and XRD patterns of electrolytic products(b)in KCl-LiCl-Li2 O(w=1%)molten salt
(3)Nd2O3、Sm2O3、Dy2O3的电解
对其他镧系元素Nd、Sm、Dy的氧化物,采用与Gd2O3相同的电极制作方式和电解条件,在LiCl-KCl-Li2O(w=1%)熔盐中进行电解。Nd2O3、Sm2O3、Dy2O3在923 K电解25 h得到的电流曲线示于图6(a)、(c)、(e)。从图6(a)、(c)、(e)可以发现:和Gd2O3的电解过程相似,均是开始时电流较大,随后降低;与Gd2O3相比,Dy2O3电极的电流变化较为频繁,可能是电极的导电性或者离子扩散性能随着电解过程发生较大变化。造成这一现象的可能原因是:(1)由表1的数据可知,Dy2O3的理论分解电压较高,电解难度较大,表现为还原电流的绝对值最小;(2)Dy2O3的最终还原率较低,说明电解中产生的金属量较少,使得电极的导电性较差,也可能导致了电流的变化频繁。与Gd2O3相比,Nd2O3和Sm2O3的电流曲线变化相对平缓,这可能是由于Nd2O3和Sm2O3阴极的电解反应速率相对Gd2O3较慢造成的。从电解电流数值来看,Nd2O3和Sm2O3阴极的初始还原电流远低于Gd2O3阴极,而它们的最终还原率相差不大(见下文)。电解反应速率较快导致O2-的消耗加大,而O2-向电极表面扩散是决定反应速率的关键步骤,所以Gd2O3阴极的电流值变化幅度较大,而Nd2O3和Sm2O3阴极的电流相对平缓。Xu等[6]的研究中也发现类似现象,在CaCl2-NaCl体系中电解Nb2O3时,当提供足够浓度的O2-时其电解电流值较大,且能够在较长的时间内保持高位稳定。图6(b)、(d)、(f)分别为相应电解产物的XRD图谱。从产物的XRD谱图中可以发现,除了夹杂的盐和中间产物外,均有很明显的金属单质相生成。而Dy2O3的XRD谱图中没有中间产物,但存在着较多的未电解的Dy2O3本体,可能因为Dy2O3的分解电压相对其它氧化物较高,所以较难被电解还原。

图6 Nd2 O3、Sm2 O3、Dy2 O3在KCl-LiCl-Li2 O熔盐中的电流-时间关系曲线(a、c、e)和相应电解产物的XRD分析图谱(b、d、f)Fig.6 Plot of i vs t(a,c,e)of Nd2 O3,Sm2 O3 and Dy2 O3 and XRD patterns(b,d,f)of electrolytic products in KCl-LiCl-Li2 O molten salt of cathode
综合以上分析,可以推测稀土氧化物RE2O3(Gd2O3、Nd2O3、Sm2O3、Dy2O3)在电解过程中可能发生如下反应(式(15)—(20))。
阴极反应:


阳极反应:
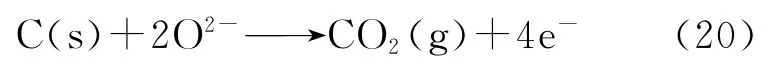
将这几种稀土氧化物(Gd2O3、Nd2O3、Sm2O3、Dy2O3)的电解产物进行氧含量分析,根据氧化物电极在电解前后的氧含量变化,计算相应的还原率分别为43.7%、40.2%、45.6%、30.5%,所得到产物的还原率较低。根据陈华林等[21]的PRS模型,在理想条件下氧化物阴极的最优孔隙率和最短电解时间计算公式如式(21)、(22)。

其中:R为金属/氧化物的摩尔体积比即V*m,金属的摩尔体积;V*0,金属氧化物的摩尔体积,对于指定金属/氧化物,R为常数;S为阴极体积的收缩率,从图5(a)插图发现Gd2O3电解前后体积几乎没有变化,而Nd2O3、Sm2O3、Dy2O3电解前后的体积也没有变化,所以S=0;M,氧化物的相对分子质量;ρ,氧化物的密度;D,O2-在KCl-LiCl中的扩散系数;c,Li2O在KCl-LiCl中的平衡浓度,在本次实验中可以假设c为Li2O完全溶解在熔盐中的浓度,c=5.12×10-4mol/cm3;m′,单位表观面积负载的氧化物的质量,本实验中稀土氧化物阴极质量为1.2 g,直径d=15 mm,厚度l=2 mm,所以m′=0.88 g/cm2;z,稀土离子的转移电子数,本实验中z=3。根据以上信息,最终可以得到Gd2O3、Nd2O3、Sm2O3、Dy2O3的最优孔隙率及其最短电解时间,结果列入表2。

表2 稀土氧化物的摩尔体积比、最优孔隙率和最短电解时间Table 2 Molar volume ratio,optimum porosity and minimum electrolysis time of rare earth oxides
据式(22)可以得到当电解反应时间为25 h,Gd2O3、Nd2O3、Sm2O3、Dy2O3最优孔隙率分别为18.7%、24.2%、30.6%、16.7%时对应的m′值,进而获得稀土氧化物(Gd2O3、Nd2O3、Sm2O3、Dy2O3)还原为金属单质的理论还原率分别为55.7%、55.7%、59.1%、54.5%,可以看出本工作的还 原 率 实 验 值(43.7%、40.2%、45.6%、30.5%)明显低于理论还原率。以Gd2O3为例,Gd2O3阴极片的孔隙率(49.9%)远高于最优孔隙率(18.7%),最后得到的还原率实验值低于理论值,这说明并不是阴极的孔隙率越高,相对应的氧化物还原率就越高。而且本实验中各稀土氧化物的电解时间也远低于其完全电解还原所需的理论最短电解时间(119~157 h)。基于以上原因,所得到的稀土氧化物还原率较低。因此,在后续研究中将采取调节氧化物阴极的孔隙率以及延长电解时间等措施对稀土氧化物的电解还原进行深入研究。
3 结 论
通过电化学循环伏安法和计时电位法研究了923 K下Li2O(w=1%)在KCl-LiCl熔盐中的电化学行为,并计算了O2-的扩散系数(D),得到D=0.5×10-5cm2/s;随后,在923 K下的KCl-LiCl-Li2O(w=1%)熔盐中,采用3.40 V的恒电压电解25 h,可以将稀土氧化物Gd2O3、Nd2O3、Sm2O3、Dy2O3部分还原为相应的金属单质,并计算了相应还原产物的还原率;同时引入PRS模型分析稀 土 氧 化 物Gd2O3、Nd2O3、Sm2O3、Dy2O3电解还原的最优孔隙率分别为18.7%、24.2%、30.6%、16.7%,最短电解时间分别为133、157、143、119 h。
以上研究结果为熔盐体系中稀土氧化物直接电解还原的进一步研究提供了理论指导和实验基础。
