嵌合抗原受体T细胞(CAR-T)药品信息可及性的国内外对比与启示
2022-11-04孙阿宁
孙阿宁
首都医科大学附属北京天坛医院药学部
张伊楠
首都医科大学附属北京天坛医院药学部
赵志刚*
首都医科大学附属北京天坛医院药学部
近年来,肿瘤免疫治疗发展迅速,已成为继手术、放疗、化疗的第四大治疗恶性肿瘤的模式。由于肿瘤免疫治疗能够提供持久的缓解,且部分晚期癌症患者对此通常具有良好的耐受性,其发现及发展已成为全球癌症疗法的重要里程碑。其中,嵌合抗原受体T 细胞(chimeric antigen receptor-T cell, CAR-T)在血液系统恶性肿瘤领域取得了显著的疗效[1],已成为学术界及产业界共同关注的焦点。CAR-T 治疗过程主要是通过从患者体内分离的T 细胞,经过体外基因修饰、扩增,再回输到患者体内,这些T 细胞利用表达的CAR 受体特异性识别并结合肿瘤相关抗原后,为T 细胞激活和增殖提供信号并释放大量细胞因子,从而发挥肿瘤免疫作用[2]。
药品信息服务能使公众通过公开的药品信息了解药品性质、用途、使用方法和注意事项,帮助医生和患者全面了解新型药品,因此保障药品信息的可及性至关重要。尤其是对于肿瘤免疫治疗这类新型药品治疗方法,个性化定制的CAR-T 类药品,不同于化学药品和一般生物制品,其产品制备方法、体内生物学特性、临床应用方式、安全性风险和价格等方面均与普通药品有显著差异,提供公众获取信息的途径有助于药品的合理应用。美国和欧盟已建立了较为完善的药品监管体系,包括对药品信息的披露。近年来我国不断完善药品监管体系,药品监管部门对已上市药品进行信息披露,但存在披露信息相对较少的问题。本文通过概述CAR-T 类药品的国内外研发现状,查询并分析美国、欧盟、日本和中国已上市的CAR-T 类药品信息的可及性,比较各国披露的信息,为进一步完善我国CAR-T 类药品的信息可及性和管理体系提供支持与参考。
1 CAR-T 类药品的临床试验及上市进展
CAR-T 产品研发迅速,目前全球已注册的CAR-T 临床试验共计1087 项[3](数据来源ClinicalTrials 官网, 时间截至2022年8月20日)。其中,中国579 项,美国366 项,欧洲93项,日本16 项。中国的CAR-T临床试验数量为世界第一,这也是中国首次在新药研发领域走在国际前列。众多临床试验结果显示,CAR-T 疗法不仅在白血病、淋巴瘤、多发性骨髓瘤等血液肿瘤的治疗中疗效卓越,其在肺癌、胃癌、卵巢癌、胰腺癌、结直肠癌等实体肿瘤也具有深厚的治疗潜力[4-5]。
CAR-T 作为目前最受瞩目的细胞免疫疗法,全球已有8 个药品成功上市(表1)。其中,国外有6 个CAR-T 产品获批上市,4个为靶向CD19 的自体CAR-T产品、2 个为靶向B 细胞成熟抗原(B cell maturation antigen,BCMA)的CAR-T 产品。 中国截至目前已有2 个CAR-T 产品获批上市,均为靶向CD19 的自体CAR-T 产品。 已上市CAR-T类药品主要用于白血病、淋巴瘤、骨髓瘤等血液肿瘤。
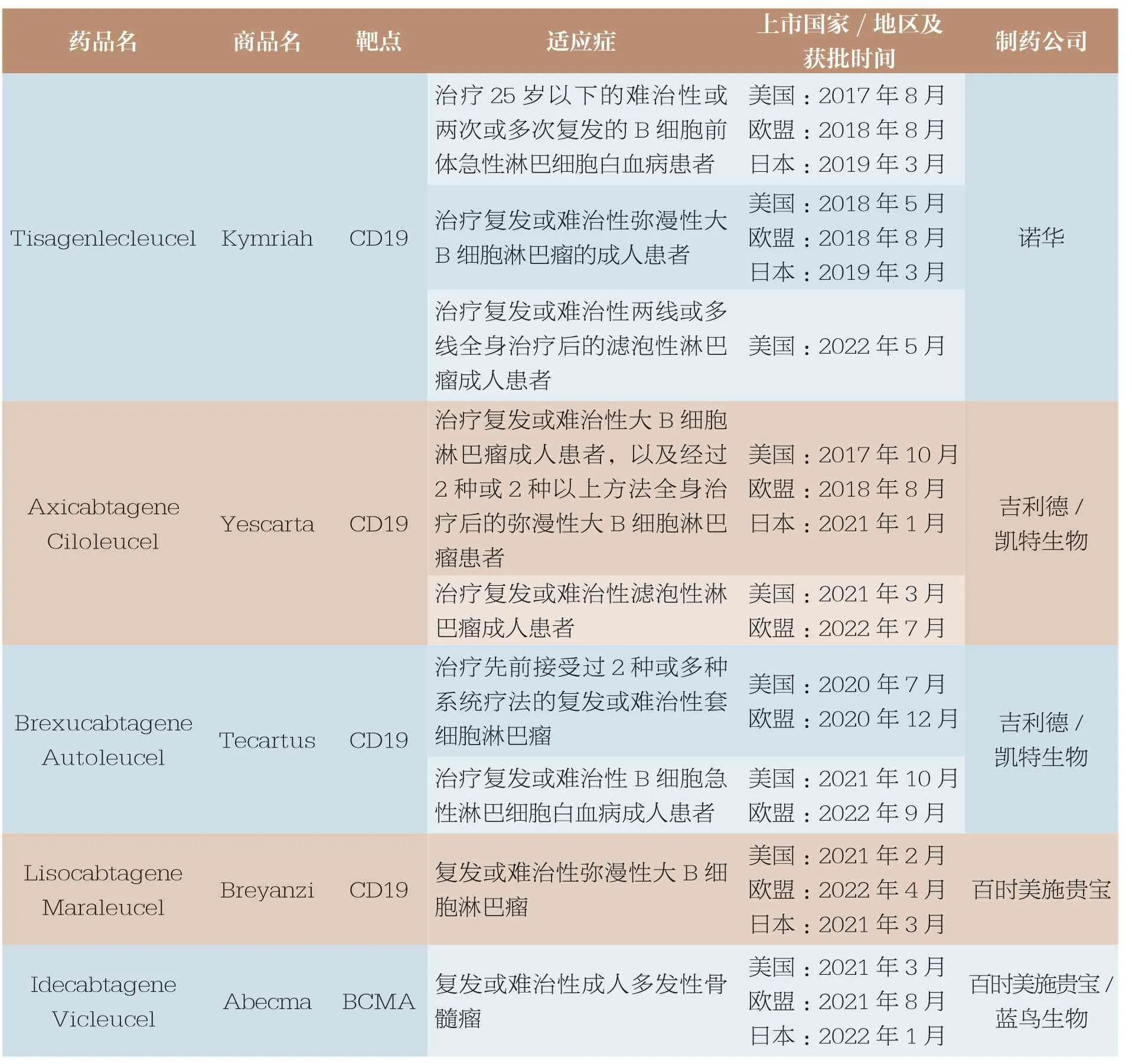
表1 全球已上市CAR-T 产品信息
2 代表性国家/地区已上市CAR-T 类药品的药品信息可及性
2.1 美国
美国食品药品监督管理局(Food and Drug Administration,FDA)的生物制品审评与研究中心(Center for Biologics Evaluation andResearch,CBER)负责管理生物制品以及血液、疫苗、细胞治疗、基因治疗和组织等相关产品。在批准的细胞和基因治疗产品页面可以查询到在美国已上市的6 个CAR-T 类药品的相关信息[6]。 每个CAR-T 类药品均有药品基本信息、产品信息和支持文件。产品信息包括药品说明书和人口统计学亚组信息。支持文件包括风险评估和缓解策略(risk evaluation and mitigation strategies,REMS)、统计审查、临床审查和评估、临床药理、批准的信件、评论等相关文件。

续表
CAR-T 类药品属于处方药,FDA 规定处方药说明书包括三大部分:①要点(highlights):简明、信息丰富的关键处方信息总结。②目录(contents):列出完整处方信息部分的章节及子章节。③完整处方信息(full prescribing information):共17 项内容,包含安全有效使用药品所需的详细处方信息。以Kymriah 为例,可查询到药品的详细信息,包括药品基本信息、药品说明书、支持文件和相关信息[7]。FDA 基于阅读对象的不同区分了专业人士和患者使用的说明书。在专业版说明书最后部分附有患者说明书药品指南(medication guides)[8],该部分以对话的口吻和简单明了的语言风格向读者介绍药品的注意事项、可能的不良反应等信息。此外,药品信息还可以在美国国家医学图书馆(National Library of Medicine)获得[9],包括药品说明书、患者说明书药品指南、药品包装照片和说明。美国药品监管部门还要求上市公司在其网站上公开说明书、用药指导和风险管理材料,相应的信息可以在诺华、吉利德/凯特生物、百时美施贵宝、传奇生物的商业网站上获取。
2.2 欧盟
欧洲药品管理局(European Medicines Agency,EMA)对于细胞和基因产品按照人用药品的先进治疗产品(advanced therapy medical products,ATMP)进行管理。药物信息由EMA 在其网站上公布,数据库名称为欧盟公众评估报告(European Public Assessment Report,EPAR)[10]。 经EMA审批上市的药物,将发布EPAR的科学评估。EPAR 是与生产企业达成一致而编写的,概述了生产企业发布的文件、描述了EMA批准此药的程序。在商业保密信息被删除后,公布在EMA 网站上[11]。可以在EMA 网站上查找到CAR-T 类药品的信息,包括药品概述、授权详细说明、产品信息文件、审评历史报告4 个部分。其中,药品概述简介了药品的特点、适应症和批准上市信息,以及通过问答的方式回答了药品的使用、优势、风险、批准原因和不良反应管理,并附有风险管理摘要文件。产品信息文件包括产品特性摘要、生产许可持有人负责批放行、上市许可条件、药品说明书和包装说明书。包装说明书的患者或护理人员的信息(package leaflet: information for the patient or carer)即专门面向普通人群的信息页面。
2.3 日本
在日本,基于细胞及组织的产品,也称为再生医学产品(regenerative medicines)。细胞和基因上市产品由日本药品及医疗器械综合机构(Pharmaceuticals and Medical Devices Agency,PMDA)管理,在PMDA 网站的再生医学产品信息搜索页面,可查询到上市的CAR-T 类药品信息,包括药品说明书、审评报告。此外,还公布了上市公司提交的申请资料(如国外上市情况及相关临床研究信息)、最佳使用推广指南。对于具有新作用机制的药品,PMDA在审批的同时会制定最佳使用推广指南,向患者及医疗机构提供使用这些产品的要求、所需信息及注意事项。药品风险管理计划(risk management plan,RMP)是日本新药上市注册及再审查的必要文件,药企对制定风险管理计划负责,而公众可通过PMDA 订阅系统获取新药风险管理计划[12]。目前,在日本上市的CAR-T 类药品等再生医学产品尚未公布风险管理计划。
2.4 中国
我国CAR-T 类上市药品由国家药品监督管理局(National Medical Products Administration,NMPA)监管,国家药品监督管理局药品审评中心(Center for Drug Evaluation,CDE)网站已经公开阿基仑赛注射液[申报名称:益基利仑赛注射液(拟定)]和瑞基奥仑赛注射液的说明书和上市技术审评报告。但目前尚未有关于CAR-T 类药品的风险管理文件、患者用药信息等支持文件,不利指导于专业人员掌握临床合理用药的核心信息和患者对药品的正确认识。
以Kymriah 为例, 上市各国的药品监管官方网站上已公开的主要药品信息见表2,因Kymriah 未在中国上市,中国以阿基仑赛注射液为例。

表2 Kymriah/阿基仑赛注射液上市国家药品监管网站上已公开的主要药品信息
2.5 美国与欧盟CAR-T类药品的风险管理
美国和欧盟上市的CAR-T类药品除了提供药品说明书、患者用药信息、审评报告,还提供了风险管理文件。美国为REMS,REMS 的目的主要是为了降低细胞因子释放综合征和神经毒性的风险,其中提出了针对发放CAR-T类药品的医疗机构、接受治疗的患者以及药品公司的要求。在审批新药申请时,FDA 会根据预计用药人群规模、所治疗疾病或症状的严重性、用该药品治疗的预期或实际持续时间、对药品效益的期望、已知的或潜在的不良事件的严重性和发生率、药品是否是新分子实体等要素来确定企业是否需要提交1 份REMS 文件,用以进行药品的风险管理。CAR-T 类药品的REMS 材料见表3,主要包括REMS 文档(REMS document)、医院登记表格(hospital enrollment form)、项目培训(program training)、患者钱包卡(patient wallet card)、 知识评估(knowledge assessment)、不良反应管理指南(adverse reaction management guide)、REMS 项目网站(REMS program website)。 上市公司制定了一系列材料来帮助完成REMS,授权代表需要完成医院登记表格并提交给REMS 项目;授权代表以及所有处方、调剂、用药相关的员工需要接受REMS 项目培训、成功完成知识评估、了解不良反应管理指南,并有相应程序确保涉及的新员工接受培训和知识评估;在患者接受治疗后出院前,应收到患者钱包卡。FDA 要求上市公司建立并维护REMS 项目网站,包含所有的REMS 材料以及处方信息、用药指南等。
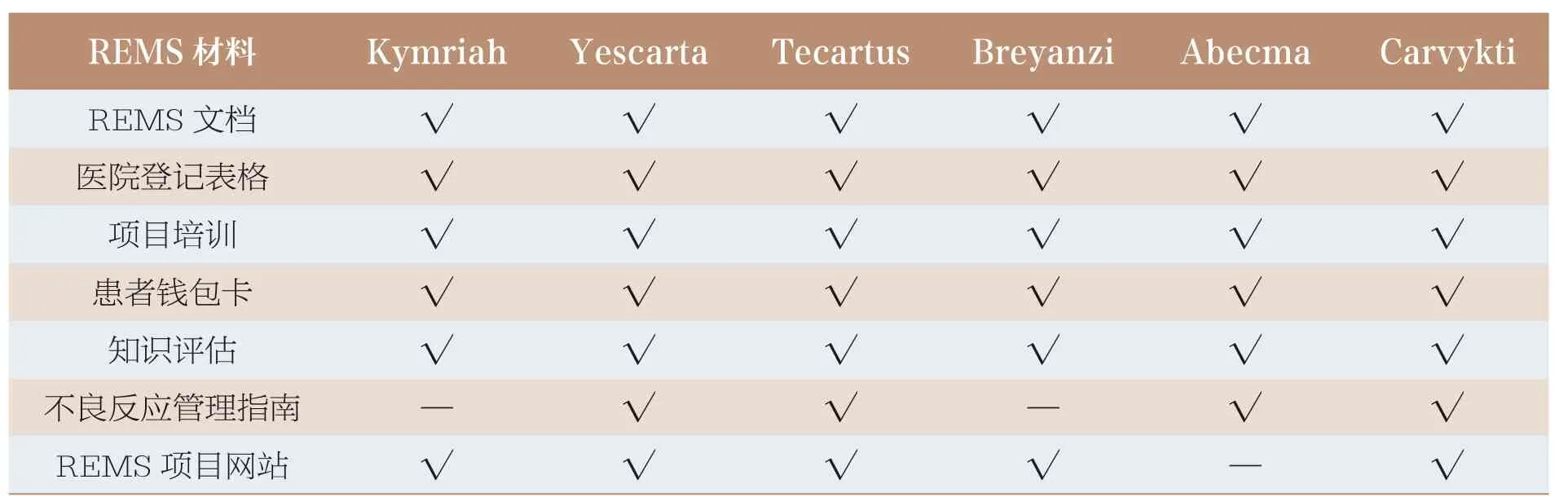
表3 美国CAR-T 类药品的REMS 材料
欧盟要求企业建立药品风险管理体系(为一系列药物警戒行动和识别、描述、预防或者最小化风险的干预措施),制定风险管理计划对其建立的风险管理体系进行描述,以识别、了解、控制药品上市后风险。欧盟要求企业提交风险管理计划作为药品审批以及评价内容的一部分,主要依据《药物警戒实践指南》(Guideline on Good Pharmacovigilance Practices)的第五模块。风险管理文件中列出了CAR-T 类药品的重要已识别风险如细胞因子释放综合征、重要潜在风险如继发恶性肿瘤、缺失信息如怀孕或哺乳期使用等,并列出了每条风险的药品关联证据、风险因素与人群、风险最小化方法及额外监管措施。
3 对我国CAR-T 类药品信息可及性管理的启示
3.1 完善药品信息相关法规和指导原则
CAR-T 治疗研发迅速,作为一种“活”的药品,给我国药品质量控制和监督管理体系带来了前所未有的挑战[17-19],其既有生物制品的属性,同时又有多样性、异质性、发展性等特性[20]。相对于传统药品,CAR-T 类药品具有研发技术含量高、技术更新迭代快、产品有效期短、制备操作环节多、质量控制难度高且要求严格、个性化程度高、对临床医生的协同要求高等特点,这对企业、医疗机构以及监管部门都提出了更高的要求和新的挑战。我国针对细胞治疗产品的监管发布了一系列相关文件,包括《免疫细胞治疗产品药学研究与评价技术指导原则(试行)》《基因修饰细胞治疗产品非临床研究技术指导原则(试行)》《免疫细胞治疗产品临床试验技术指导原则(试行)》等。药品信息的法制化管理涵盖药品研究、生产、流通、使用等所有存在药品信息的环节,为强化广大患者的知情权益,保证公众用药安全,我国以《药品管理法》作为法律基础,出台了《药品说明书和标签管理规定》等一系列的法律法规[21]。但是在公开披露药品风险管理信息、药品上市后再评价信息、针对医务人员和患者的可读性信息等方面需要制定相应的法规和规范。建议对药品信息实行全方位监管,建立贯穿免疫细胞治疗产品制备、药学研究、临床研究、注册申报、药品风险管理、上市后再评价的一系列法规文件,规范药品信息公开,促进药品研发人员的药物研究与开发,促进医疗机构人员获得准确的药品信息,提高临床使用的合理性,以提高免疫细胞治疗领域的规范和快速发展。
3.2 提高我国药品信息可及性
目前在美国、欧盟、日本和中国上市的CAR-T 类药品均可以在上市国家药品监管网站上查找到药品的最新药品说明书和上市审评报告。FDA 和EMA 网站上药品信息丰富,科研人员可获取药品研发信息,医生可获取临床使用信息,药师可获取临床研究信息,公众可获取通俗易懂的药品信息。美国和欧盟按照各自执行的法规,要求药品公司制定风险评估和缓解策略、风险管理计划并执行,风险管理文件均在药品监管网站公开。日本药品监管网站公布了上市公司提交的申请资料,以及最佳使用推广指南。我国CDE 目前公开的药品信息为药品说明书和上市审评报告。其中,药品说明书只包含了专业内容,缺少适合患者阅读的信息页面,不利于药品的安全合理使用以及公众对于CAR-T 类药品的正确认识。建议我国官方平台对于国内已上市的CAR-T 产品,在及时公开药品说明书和技术审评报告的基础上,增加针对不同群体用药指导,使医生、患者和公众全面了解药品信息;增加风险管理文件,以提高医务人员及患者正确用药、及时采取安全对策;可要求上市公司在其网站上公开药品说明书、用药指导和风险管理材料,以增加药品信息获取途径,提高公众对药品的正确认识。

3.3 完善药品风险管理体系
CAR-T 疗法在临床治疗过程中经常伴随不同的不良反应,尽管通过相应的支持治疗,这些毒性通常是可控制和可逆的,但仍存在危及患者生命的严重不良反应的风险,需要密切警惕和及时治疗。CAR-T 的毒性作用分为CAR-T 相关的毒副作用、非肿瘤靶向作用以及长期毒副作用,常见的由T 细胞激活和扩增导致的毒副作用有细胞因子释放综合征和免疫效应细胞相关神经毒性综合征[22]。Kymriah 说明书中列出的严重不良反应还包括感染和发热性中性粒细胞减少症、长期血细胞减少症、低球蛋白血症。Abecma 的黑框警告还包括噬血细胞性淋巴组织细胞增多/巨噬细胞活化综合征和伴有出血和感染的长期血细胞减少症。因此,正确有效地对CAR-T 相关不良反应进行管理和干预,已成为CAR-T 免疫治疗成功的关键步骤。
美国和欧盟均披露了CAR-T 类产品的风险管理文件,我国《药品管理法》中明确指出药品上市许可持有人应当制定药品上市后风险管理计划等要求。CDE 先后发布《风险分析与管理计划撰写指导原则(征求意见稿)》《“临床风险管理计划”撰写指导原则(试行)》。2022年1月26日,为规范和指导CAR-T 治疗产品申请上市注册时风险管理计划的撰写,CDE 组织制定了《嵌合抗原受体T 细胞(CAR-T)治疗产品申报上市临床风险管理计划技术指导原则》。但目前我国尚未将风险管理文件正式规定为新药上市注册申请材料,也未作为公开资料应用于药品上市后风险的管理。因此,我国需明确规定企业提供风险管理文件,完善我国药品风险管理的法规和指南,规范公开披露风险管理文件,从而有效控制风险,最大限度地保障患者用药安全。
