药品上市许可持有人制度下的偏差处理和变更控制探讨
2022-11-04刘超
刘超
浙江省药品化妆品审评中心
罗英
浙江省食品药品检验研究院
付莉娜
浙江省药品化妆品审评中心
卜华荣
浙江和泽医药科技股份有限公司
周耘*
浙江省药品化妆品审评中心
药品上市许可持有人(MAH)制度采用的是上市许可与生产许可分离的管理模式。我国的MAH 制度自实施以来,解除了药品注册和生产许可的捆绑模式,极大地激发了研发机构的积极性,使得医药行业创新活力得到有力释放,并且在专业分工、资源配置、重复建设、监管效能上都有很大的改善和提升。MAH 制度改变了药品管理和责任承担的机制,MAH 将承担药品整个生命周期内质量与风险的主要责任,这对MAH 的责任和管理能力提出了更高的要求,同时给药品监管带来了新的挑战。MAH 制度下,上市许可持有人和生产许可持有人可以是同一主体,也可以是两个相互独立的主体,其允许MAH自行生产药品,也可以委托其他生产企业进行生产。在委托其他生产企业生产药品时,MAH 如何履行责任并切实做好风险管理工作成为现实难题。作为《药品生产质量管理规范》(GMP)的重要组成部分,良好的偏差处理和变更控制是确保药品质量的重要措施之一。本文通过分析国内外MAH 管理以及监管部门对偏差处理和变更控制监管现状,为我国MAH 制度下的偏差处理和变更控制提出建议与对策,为监管部门提出监管政策建议,同时为MAH 加强质量风险管控体系建设、进一步提升药品质量管控能力提供参考。
1 国内外MAH 管理现状
1.1 美国
美国是施行MAH 制度最早的国家之一。《联邦食品药品和化妆品法案》(Federal Food, Drug and Cosmetic Act,FD&C Act)使用申请人(applicant)和申请持有人(applicant holder)来表示药品上市许可申请人(MAA)和药品上市许可持有人(MAH)[1-2]。申请人和申请持有人均是药品申请或者产品上市的责任主体,并且规定企业负责人是质量第一责任人。
1.2 欧盟
欧洲经济共同体在其1965年颁布的Directive 65/65/EEC[3]指令提出实施MAH 制度,规定药品批准证明文件的所有者(MAH)要承担药品全生命周期的管理主体责任。欧盟现行的MAH 制度既考虑了整体性,又兼顾了成员国的自主性。根据欧盟各国实际状况,可分为药品上市许可申请人(MAA)、药品上市许可持有人(MAH)和药品生产许可持有人(PLH)三种责任主体,分别承担在药品批准上市前、批准上市后批文持有者、药品生产或受托生产实体的三种责任,现行 的Directive 2001/83/EC[4]沿用了MAA 和MAH 的定义。
1.3 日本
2005年日本修订生效的《药事法》[5]正式引入MAH 制度,与欧盟和美国相比较晚,却也是亚洲较早实施MAH 制度的国家。《药事法》将生产许可和上市许可申请相互独立,与美国和欧盟不同的是日本提出了“上市许可人执照”制度,必须取得某一类MAH 执照后,才可以提出具体产品的上市申请,属于资格准入型MAH 制度。
1.4 中国
我国的MAH 制度试点在2015年拉开帷幕,2019年新修订《药品管理法》正式实施,标志着我国上市许可持有人制度全面落地。后续出台的《药品生产监督管理办法》[6]《药品委托生产质量协议指南(2020年版)》[7]等法规的相关内容都是对MAH制度实施的补充与指导。
我国MAH 制度的实施面临着如何确保药品从研发、生产、销售直至产品退市全生命周期的质量安全的挑战。同欧盟、美国、日本等国家和地区相比,我国MAH 制度实施较晚,行业积累的经验相对不足,跨境持有和跨境委托生产、境内境外主体变更相互转换尚未完全实现。基于风险考虑,特殊药品的委托生产也未完全放开,还不允许生物制品原液、制剂分段委托生产。此外,我国的药物警戒工作也刚刚起步,产品上市后安全性研究、患者赔偿保障能力相对不足,MAH 质量管理能力、风险防控能力、责任赔偿能力这“三大能力”还需要不断提高。
2 国内外监管部门对偏差处理和变更控制的监管现状
2.1 美国
美国食品药品监督管理局(FDA)发布的多部法规、行业指南,对MAH 委托生产情形,MAH 和受托方的总体责任进行了界定;推荐签订质量协议约定好各方的职责,以确保委托生产cGMP(现行药品生产管理规范)的符合性。美国卫生和公共服务部(HHS)和FDA 共同制定的行业指南《药品委托生产质量协议》(Contract Manufacturing Arrangements for Drugs: Quality Agreements)[8]规定,质量协议应描述MAH 与受托方在药品生产中各自的cGMP 相关义务、职责和活动。解释受托方如何向持有人报告生产偏差,以及如何按照cGMP 要求调查、记录和解决偏差;MAH 和受托方均可以启动对工艺、设备、检验方法、质量标准和其他合同约定要求的变更。双方应对变更进行讨论,在质量协议中对其进行说明,还应列出所有变更要如何管理,包括在执行变更之前实施所需验证活动的职责分配。但是对于哪些变更需要由MAH 在实施之前进行审核和批准,哪些变更受托方可以直接实施,《药品委托生产质量协议》中还未给出更明确的指导意见。
2.2 欧盟
欧盟与MAH 制度相关的法规主要为欧洲药品管理局(EMA)发布的《外包服务合同框架草案》[9](Draft framework service contract for outsourcing)及《外包服务的规范流程》[10](Steps involved in outsourcing of services)等, 为欧盟药品外包服务从法规层面提供了一个严谨、完整的合同样本,其涵盖从合同建立到合同执行再到合同终止的所有环节,覆盖整个药品委托加工周期,规范委托双方的行为,保证各方利益,使委托加工更具操作性。但委托生产相关规定中并未提及当发生变更和偏差时,哪些情形下由持有人批准关闭,哪些情形下由受托方批准关闭等指导意见。
2.3 日本
日本与MAH 相关的文件主要为《制造委托合同》和《关于委托生产合同签订的一些注意事项》,其中《制造委托合同》属于一个完整的委托生产的合同模板,包含了签约的目的、制造说明书、原材料等的供应、产品的生产量、分包、业务执行中的合作义务、成品检验、质量保证、提供材料的义务、支付、成本、产品运输、合同解除、无法制造、保密、合同期限、商谈事项等17 项内容。其中涉及的变更主要是指合同本身内容发生变化,例如生产数量、合同存续终止等发生变化,但对于委托生产中变更和偏差等内容并未进行明确阐述。
《关于委托生产合同签订的一些注意事项》主要阐述委托生产前,委托方和受托方签订合同中应明确的事项,文件中规定对于委托生产后出现的变更和偏差需要双方协商处理。
2.4 中国
我国2020年发布的《药品委托生产质量协议指南(2020年版)》强调MAH 的主体责任,要求偏差和变更均应由MAH 审核。MAH 作为责任主体,要按照药品监督管理部门的规定,全面评估、验证变更事项对药品安全性、有效性和质量可控性的影响。MAH 和受托方应按照相关法律法规、技术规范等开展变更。任何一方进行可能影响药品质量的变更都应及时书面告知对方。质量协议应规定双方均须建立变更控制程序,明确发生变更时的工作措施;还应规定委托生产产品相关变更的风险程度由MAH 评估确定,受托方在变更实施前需要经MAH 审核批准。此外,质量协议应规定受托方在生产质量管理活动中发生偏差时应按照偏差处理程序进行处理。受托方需评估与受托生产产品相关的所有偏差对产品安全性、有效性和质量可控性的影响,并根据偏差的性质、范围及对产品质量的影响程度实施分类管理,将拟采取的纠正预防措施报告MAH。偏差处理报告需经MAH审核批准。
2.5 小结
综上,通过比较国内外制定的外包、MAH 委托生产的流程、框架、质量协议等相关指导原则,均明确了MAH 和受托方都应该建立各自的义务和责任,但针对变更和偏差的具体事项,其他国家和地区的监管机构多持开放态度,未对双方的责任进行界定。我国则是相对强调MAH 的主体责任,但实际在MAH 制度实施过程中,委托生产过程中的变更和偏差情形较为复杂,双方对各自的责任需要更明确、更具体的指引。
3 我国MAH 和受托生产企业对偏差处理和变更控制的工作程序及策略
结合MAH 制度运行情况,本文针对我国不同机构、不同岗位人员进行问卷调查,共收集有效问卷131 份。调查问卷按不同机构划分,监管机构/高校/事业单位共占7.63%,自行生产的药品上市许可持有人企业(持药品生产许可证A 证)占61.83%,委托生产的药品上市许可持有人企业(持药品生产许可证B 证)占48.85%,接受药品上市许可持有人委托的企业(持药品生产许可证C 证)占21.37%。
3.1 变更控制
变更问题主要涵盖机构与人员、厂房与设施、设备、物料与成品、文件管理、生产管理、质量控制与质量保证、委托生产与委托检验等八方面内容,接受问卷调查者需选择每个变更问题由受托方决定、持有人决定还是由双方共同决定。根据调查结果显示受托方对于其人员管理、车间资产配置有较大的主动权;机构与人员、厂房设施、设备相关的变更倾向于由受托方决定或由双方共同决定;物料与成品管理更倾向于由MAH 决定,其他的变更事项支持双方共同决定,调查结果详见表1。
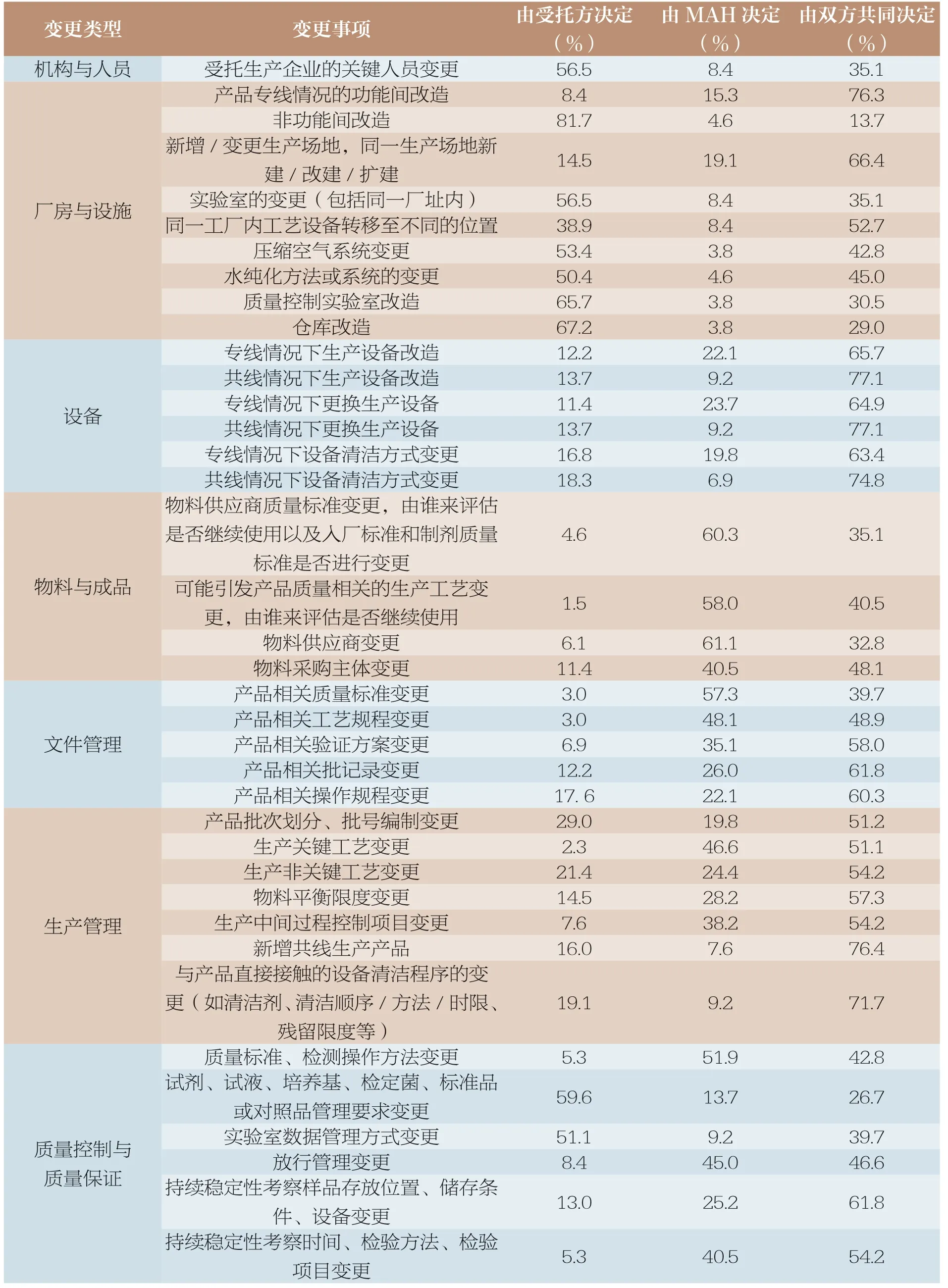
表1 变更控制调查结果

续表
3.2 偏差处理
偏差问卷调查中对应其严重程度分为4 档,1~3 分为一般,4~5 分为较严重,6~8 分为严重,9~10 分为极为严重。接受调查者需要给所有偏差问题进行打分,并且还需要给出几分以下可由受托生产企业自行关闭的建议。
本文偏差共设置17 项问题,收集到的严重程度评分均为4 分以上,结果详见图1。
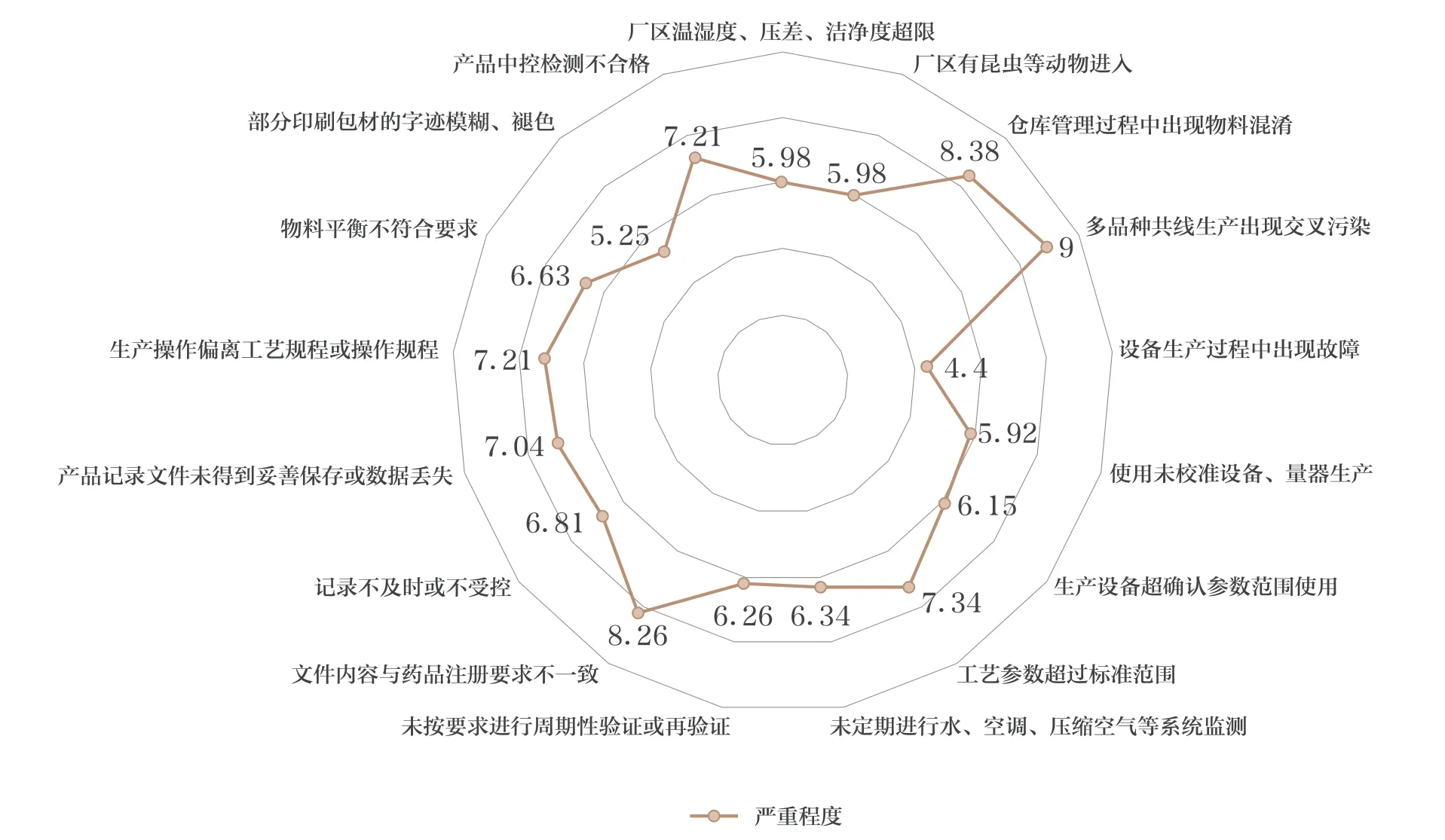
图1 偏差处理调查分析结果
问卷结果支持4 分以下的偏差可由受托生产企业自行关闭,即大多数从业人员接受一般偏差由受托方自行关闭,较严重、严重、极为严重的偏差均需汇报给MAH。由此可知从业人员对于偏差的处置较为谨慎。
3.3 监管主体
根据监管职责的调查结果显示:对于偏差处理的监管,接受调查者认为应由受托生产企业所在地药品监管部门监管的占87.02%,这与偏差本身主要发生在生产活动过程中直接相关。而对于变更控制的监管,尽管MAH是药品上市后变更的责任主体,但接受调查者认为应由MAH 所在地药品监管部门监管的仅占35.11%,这可能是考虑到受托生产企业是最终实施者,而MAH所在地药品监管部门难以履行监管职能。
4 建议与对策
基于问卷调查的结果,通过专家专题研讨论证,对偏差处理和变更控制的管理职责,本文最终形成了以下几点建议。
4.1 变更控制策略
对于变更控制的执行,本文按 8 个类型的40 个事项,分别给出了建议的控制策略,如受托生产企业的关键人员变更最终应由受托方决定,而产品专线情况的功能间改造由双方共同决定,具体建议详见表2。
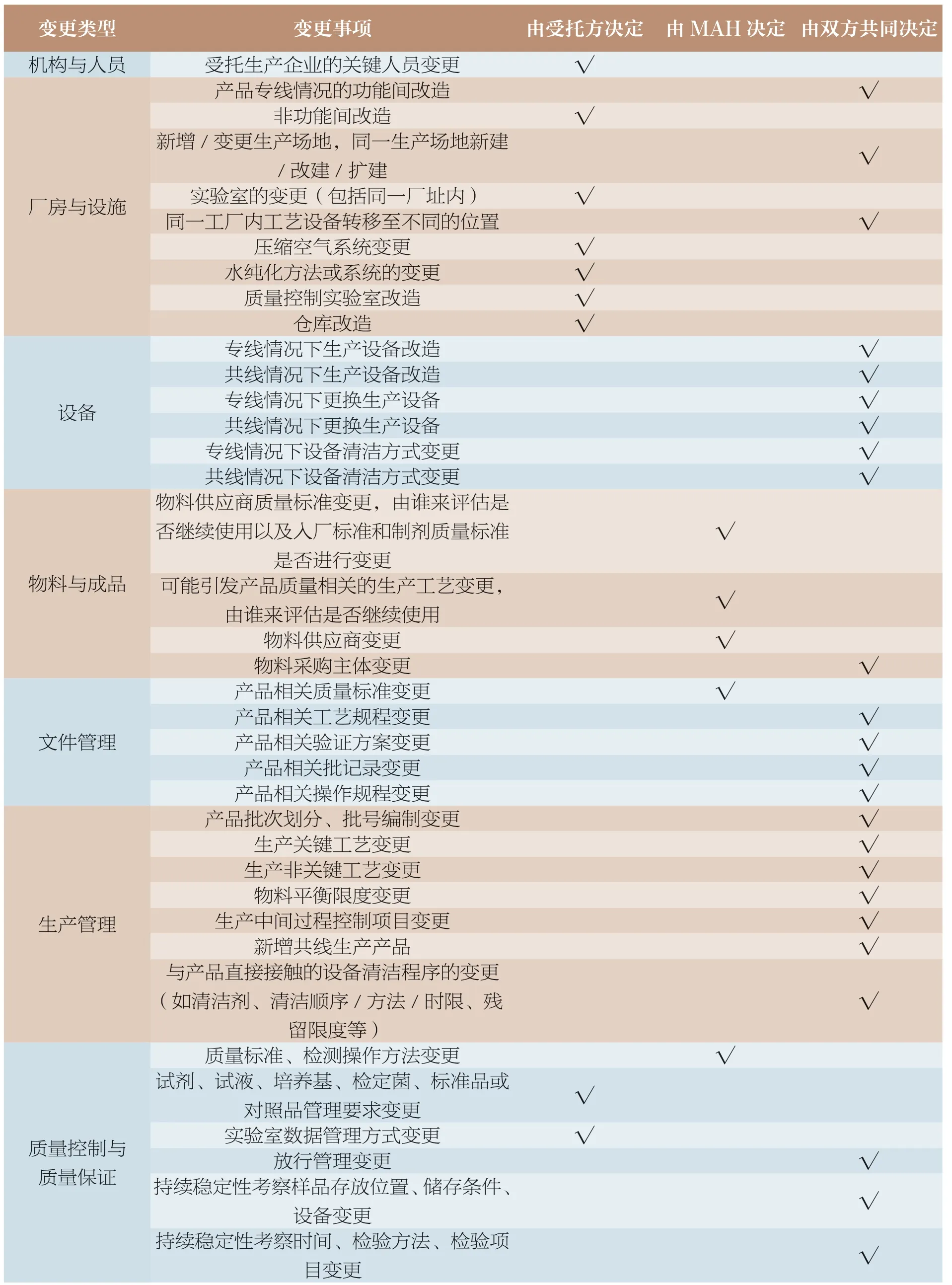
表2 变更控制策略

续表
4.2 偏差处理策略
MAH 和受托生产企业双方对于偏差的处置都较为谨慎。因此,建议与MAH 委托生产的品种相关的偏差发生时,均应及时汇报给MAH,由MAH 评级。对于不影响产品安全性、有效性和质量可控性的微小偏差,由受托方进行记录、调查、评估和跟踪。在产品放行时,MAH 应当对所有偏差进行审核。对于可能影响产品安全性、有效性和质量可控性的偏差和检验结果偏差,受托方需要报MAH 审核批准。
4.3 监管展望
MAH 作为药品上市许可的责任主体,承担药品全生命周期的安全性有效性保证义务,必须有质量管理能力、风险防控能力、责任赔偿能力。MAH 和受托生产企业应以风险管理为原则,采取有效措施对偏差和变更进行处理和控制。MAH 制度的实施,使得药品上市后监督管理措施更加有力。对于不在同一区域的MAH 与受托生产企业,可采取互联网+检查、联合监管等模式。在具体措施上,通过引入约谈、告诫信、限制使用、召回等机制,根据双方质量协议中的责任划分,对MAH 和生产企业追责,同时追究相关责任人责任。
