Gasdermin 家族蛋白介导的细胞焦亡及其病理作用与靶向干预
2022-10-21白杨邓万燕闵睿刘星
白杨,邓万燕,闵睿,刘星
(1. 中国科学院上海巴斯德研究所微生物、发育与健康研究中心,上海 200031;2. 中国科学院大学,北京 100049;3. 上海华申微生物与感染研究所,上海 200052)
细胞焦亡是一种程序性的炎性细胞死亡,其在启动机制、形态与分子学特征以及参与的生理病理过程方面都与细胞凋亡存在显著差异。凋亡参与调控机体发育和衰老细胞的清除,凋亡过程一般不会释放细胞内容物或引起机体的炎症反应;而焦亡一般发生于病原感染等病理过程中,伴随着细胞膜的破裂和内容物释放,具有强烈的促炎效应。最近的研究发现,gasdermin(GSDM)家族蛋白是细胞焦亡的直接执行者,该家族蛋白一般具有高度保守的N 端和C 端结构域,N 端结构域大多能够靶向细胞膜并具有膜成孔活性,能够直接触发细胞死亡及随后的细胞内容物释放;而静息状态下C 端结构域可抑制膜成孔活性,因此GSDM 的激活通常依赖于焦亡启动过程中上游蛋白酶的靶向剪切活化。目前为止,人类GSDM 家族中共有6 个同源基因被发现:GSDMA,GSDMB,GSDMC,GSDMD,GSDME(也称为DFNA5)及PJVK[也称为常染色体隐性遗传性耳聋59 型基因(deafness autosomal recessive type 59,DFNB59)]。GSDM 家族的首个成员Gsdma1被发现表达于小鼠胃肠道与皮肤,“gasdermin”也因此得名。
GSDMD 是研究最多的GSDM 成员,其活性在炎症小体介导的细胞焦亡中发挥关键作用[1-3]。GSDME 可被半胱氨酸-天冬氨酸蛋白酶-3(cysteinaspartate protease-3,caspase-3)激活从而介导细胞死亡形式从凋亡向焦亡转换[4-5]。肿瘤细胞中GSDMB,GSDMC 及GSDME 的激活被发现能够促进宿主免疫系统对肿瘤细胞的清除[6-8]。最近的研究发现GSDMA 可被病原菌分泌的毒素激活并介导宿主的抗感染免疫[9-10]。而目前为止PJVK 激活机制和功能仍然未知(见图1)。
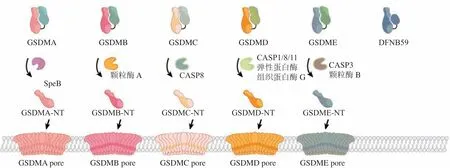
图1 Gasdermin 家族成员及其成孔活性触发机制Figure 1 Gasdermins and their pore-forming mechanisms
在GSDM 成孔触发焦亡的过程中,GSDM膜孔洞形成后损伤的膜呈大的膨胀泡状,同时许多低相对分子质量的细胞内容物如炎症因子以及危险信号分子包括腺苷三磷酸(adenosine triphosphate,ATP)和高迁移率族蛋白B1(high mobility group protein B1,HMGB1)等都通过细胞膜上的GSDM 孔洞无选择性地释放到胞外,引起组织炎性反应,由此招募免疫细胞到达相应感染或组织损伤部位[11-12]。越来越多的研究证实GSDM 介导的细胞焦亡与各种疾病发生发展之间存在复杂而密切的联系,包括脱发、哮喘、听觉损伤、感染性疾病和癌症等[13-20]。因此,靶向GSDM 家族蛋白的治疗策略开发也逐渐成为研究热点。本综述总结了GSDM 家族蛋白的表达、激活和调控机制,以及它们作为细胞焦亡的执行者在多种疾病发生发展过程中的重要功能,并讨论了以GSDM 为靶点开发新型治疗药物的前景与挑战。
1 Gasdermin 家族蛋白及其活化机制
GSDM 家族的首个成员发现于2000 年,在毛发发育缺陷的小鼠内克隆得到,后命名为Gsdma1[21-22]。鉴定发现小鼠含有3 个Gsdma同源体——Gsdma1,Gsdma2和Gsdma3,且都位于小鼠染色体11D 的同一基因簇,后两者分别主要表达于小鼠的胃和皮肤[23-24]。与其对应的人的同源基因GSDMA在胃和皮肤内均高表达。 最新研究显示,上皮细胞中的GSDMA 可被化脓链球菌(Streptococcus pyogenes)分泌的毒力因子 —— 链球菌热原外毒素B(streptococcal pyrogenic exotoxin B,SpeB)剪切激活,在感染过程中上调炎症反应并促进机体对病原菌的清除[9-10](见图1)。人GSDMB与GSDMA位于同一基因簇,而啮齿动物不表达GSDMB[25]。相对于GSDMA,GSDMB表达更广泛,主要表达于气道与胃肠道上皮细胞、肝脏细胞、神经内分泌细胞以及免疫细胞[16,26-27]。研究显示GSDMB 可被来自细胞毒性T 细胞或自然杀伤细胞的颗粒酶A 剪切活化并进而诱导焦亡[7](见图1)。另外,已发现GSDMB有4 种不同的剪接异构体,但这些异构体的表达是否存在组织或细胞特异性及其功能是否存在差异仍然未知。GSDMC最初被发现在转移性黑色素瘤中异常高表达,因此被命名为黑色素瘤衍生的含亮氨酸拉链的核外因子,并作为黑色素瘤发展的分子标记[26,28-29]。与人源GSDMC相对应,小鼠有4 个同源体Gsdmc1-4。GSDMC主要表达于气管、脾脏、食管、小肠、盲肠、结肠等组织[26,28,30],也有研究发现其表达于皮肤角质细胞[31-32]。最近研究发现GSDMC 可被caspase-8 剪切激活[8](见图1)。GSDMD基因位于人类8 号染色体临近GSDMC的位置[29],在免疫细胞(尤其是巨噬细胞和树突状细胞)、胎盘、胃肠道上皮细胞皆有表达,此外还表达于多种癌细胞系[26,29,33-34]。GSDMD 介导的细胞焦亡途径近年来已被广泛研究,GSDMD 可被炎症小体通路中的炎性caspase 激活。炎症小体是胞内一类响应危险信号后组装形成的多蛋白复合物,可分为经典和非经典2 种途径。经典炎症小体能够响应不同刺激,进而通过激活caspase-1 来介导GSDMD 的剪切;在非经典炎症小体途径中,进入胞质内的脂多糖(lipopolysaccharide,LPS)能够直接激活炎性caspase-4/5/11 剪切GSDMD[1-2]。耶尔森菌(Yersinia)产生的效应因子耶尔森菌外部蛋白J(Yersiniaouter proteins J,Yop J)能够引起宿主细胞内caspase-8依赖的GSDMD 剪切,该过程依赖于Yop J 对宿主转化生长因子β 激酶1 活性的抑制[35-36]。此外,GSDMD 还可被其他蛋白酶如弹性蛋白酶和组织蛋白酶G 剪切激活[37-38](见图1)。GSDME 最早被发现其突变会导致家族遗传性耳聋,其转录本在耳蜗、胎盘、心脏、大脑和肾脏中均被检测到[17,39]。GSDME除了能被caspase-3 剪切介导凋亡向焦亡的转换之外[4-5],还能在肿瘤免疫发生过程中被颗粒酶B 剪切激活并诱导肿瘤细胞的焦亡[6](见图1)。PJVK编码了GSDM 家族中与其他成员同源性较低的蛋白,其C 端结构域显著短于其他成员。在大脑、眼、内耳、心、肺、肾、肝、肠和睾丸中均可检测到PJVK转录本[40-41]。人PJVK突变引发的病理表型包括耳蜗外毛细胞功能失调[42]以及听觉神经病变[40]。通过正向遗传筛选发现小鼠的Pjvk突变也和耳聋存在相关性[42],但至今为止PJVK 是否具有成孔活性及其激活方式仍不清楚。
2 Gasdermin 家族蛋白的成孔机制与效应
2.1 Gasdermin 孔洞的结构特征与形成机制
大多数GSDM 家族蛋白都由3 个基本结构域组成,包括具有成孔活性的N 端结构域(N-terminus,NT),又称为成孔结构域;抑制N 端成孔活性的C端结构域(C-terminus,CT),又称为抑制结构域;连接N 端和C 端结构域之间的连接结构域(linker)。以GSDMD 为例,炎性caspase 在GSDMD 的linker区域剪切,产生1 个相对分子质量约31 000 大小的NT,以及1 个相对分子质量约22 000 大小的CT[43-44]。NT 释放后能够转位至细胞膜,并在细胞膜上多聚化形成孔洞结构,导致细胞膜失去完整性,诱发细胞焦亡[45-47]。GSDMD-NT 的细胞膜转位是由于其能直接与组成细胞膜的酸性磷脂(包括磷脂酰肌醇、心磷脂、磷脂酰丝氨酸等)产生较强结合[43-44,48-49]。GSDM 家族成员的NT 基本具有相似的脂亲和性[5,43],由此可以推测其与细胞膜结合的机制类似。
GSDM 全长蛋白及其在脂质体上形成的孔洞的首次精细结构解析来源于对小鼠GSDMA3 蛋白结构的研究[43,50]。结构分析发现GSDMA3-NT 的核心结构为9 个β 股扭曲延伸形成的β 折叠,周围存在若干个α-螺旋结构;GSDMA3-CT 的中心则为多个α螺旋形成的紧密球状结构,外部由3 股较短的β 折叠覆盖(示意图见该处引用文献的Figure 4a)[43]。当GSDMA3 蛋白未被剪切时,其CT 与NT 的α1和α4 螺旋分别产生非共价结合,这一相互作用将GSDMA3 维持在自抑制的状态;当GSDMA3 的连接结构域发生剪切,这种自抑制便会解除[43]。与处于自抑制状态时相比,NT 被释放活化之后在构象上发生较大改变[50]:原本埋入蛋白内部的α 螺旋和β 折叠伸展出来,β3-β4-β5 和β7-α4-β8 分别形成了2 个发卡结构,从NT 的核心球状区域向外延伸。此时整个NT 的结构类似左手,其核心球状结构域形似手掌,2 个插入膜结构的发卡结构则类似手指。这些结构变化同时产生了磷脂结合界面和寡聚化界面:带正电的α1 螺旋形状类似横在手掌前的大拇指,是主要的磷脂结合区域,2 个发卡结构则类似并拢的手指插入膜磷脂内;而寡聚化由其手部构象的边缘介导,从穿膜的手指部分到折叠的拇指部分再到手掌。尤其是各个单体的α1 螺旋在GSDM 孔洞中头尾对齐,形成稳定的螺旋带(示意图见该处引用文献的Figure 2a,2b)[50]。以上变化使得NT 能够在结合并插入细胞膜磷脂层的同时发生聚合,最终在细胞膜表面形成稳定的环状孔洞。该孔包含26 ~28 个单体,孔内径18 nm,外径为28 nm,可容许一些相对分子质量较小的蛋白质如细胞因子等的通过[11,43,51]。蛋白结构相关研究为GSDM 家族蛋白成孔机制提供了依据。
2.2 Gasdermin 孔洞的通透性、细胞毒性与调控机制
一般情况下,GSDM 剪切引起活性GSDM-NT的释放以及后续的膜成孔,最终会导致细胞焦亡。狭义上的焦亡指依赖于上游炎症小体和炎性caspase的激活触发的GSDMD 依赖的炎性细胞死亡过程[52]。由于GSDM-NT 本身的成孔活性并不依赖于炎性caspase,并且研究发现除炎性caspase 之外的其他蛋白酶(例如颗粒酶、弹性蛋白酶等)也能够剪切活化GSDM 蛋白[6-7,53],细胞焦亡的定义被扩展为由成孔活性的GSDM-NT 介导的细胞死亡形式[47,54]。
焦亡的发生伴随着细胞的气球状膨胀、细胞膜通透性改变以及细胞内容物的释放。虽然GSDM孔洞的形成会引起细胞膜完整性的破坏,但最终导致细胞膜解体的原因有待进一步深入探究。有研究认为该过程依赖于细胞膜内外的渗透压差异。一般情况下胞质的钠离子浓度低而钾离子浓度高,而胞外环境相反;当GSDM 在细胞膜上组装形成孔洞时,钠离子内流并携带与其水合的水分子,导致细胞体积增加,一旦体积超过膜容量,质膜就会以气球的形式膨胀并与膜附近的细胞骨架分离,随后细胞会彻底涨破。这种裂解方式可释放胞质内相对分子质量较大的可溶性蛋白质[如乳酸脱氢酶(lactate dehydrogenase,LDH)等],最终剩余的细胞结构形成细胞残骸[1,55]。最近另一项研究提出焦亡发生时细胞膜以及细胞的裂解是主动的过程,该研究发现疾病特征性神经损伤诱导蛋白1(nerve injuryinduced protein 1,NINJ1)在细胞质膜解体过程中发挥至关重要的作用。NINJ1 缺失的巨噬细胞在细胞焦亡发生过程中表现出细胞膜通透性改变(ATP等相对分子质量较小的细胞内容物发生外流),但相对分子质量较大蛋白(包括HMGB1 和LDH)的释放受到抑制,焦亡细胞一直保持气球状形态[56]。另外,Ninj1敲除小鼠比野生型小鼠更易受柠檬酸杆菌的侵害,这也表明细胞膜是否彻底破裂对于宿主防御病原入侵非常关键[56]。
在感染引发的细胞焦亡过程中,促炎症因子白细胞介素(interleukin,IL)中IL-1β 和IL-18 前体在经过炎性caspase-1 剪切成熟后,能够通过GSDM产生的细胞膜孔洞释放到胞外[43-44]。因此GSDM 孔洞能够辅助这类由于缺乏分泌肽而无法通过经典分泌途径释放的细胞因子分泌到胞外,持续性地产生免疫效应,引起机体的炎症反应。蛋白质组学分析鉴定发现焦亡过程还伴随了900 余种细胞蛋白质分子释放至胞外[57-58],其中相当一部分在胞外发挥的作用目前仍不明确。除了相对分子质量较小的蛋白质,GSDM 孔洞还可介导离子的非选择性流动,影响细胞死亡的进程:在非经典炎症小体通路中被进入胞内的LPS 激活后,GSDMD 活化成孔引发胞内钾离子外流,随后导致经典炎症小体NOD 样受体热蛋白结构域相关蛋白3(NOD-like receptor thermal protein domain associated protein 3,NLRP3)通路的激活,加速细胞发生焦亡[59-60];嗜肺军团菌(Legionella pneumophila)感染过程中也存在类似机制[61]: 在黑色素瘤缺乏因子 2 炎症小体激活时, GSDMD 活化导致的钾离子外流抑制了胞质DNA 起始的环磷酸鸟苷-腺苷合成酶依赖的Ⅰ型干扰素诱导通路[62]。
此外,焦亡过程中GSDM 可能同时在其他细胞器膜上成孔,进一步调控细胞焦亡进程。前文提到,体外实验证明GSDM-NT 可与心磷脂结合[43-44]。由于心磷脂是构成哺乳动物细胞线粒体膜的磷脂组分之一 ,GSDM 家族有可能靶向线粒体并发挥作用。最近的研究发现GSDMD 和GSDME 的NT能转位至线粒体并使其外膜通透性改变,引发活性氧的产生、细胞色素c 的泄露以及线粒体膜电位丧失[63-64]。此外,研究发现GSDMA3-NT 可以破坏线粒体并引起线粒体自噬[65]。但GSDM-NT 对线粒体的靶向是否由心磷脂介导有待进一步探究。除了作为细胞焦亡的关键执行分子,中性粒细胞GSDMD 被活化后可结合并使核膜与细胞膜通透性改变,引起中性粒细胞基因组DNA 的释放,释放的DNA 可作为中性粒细胞胞外诱捕网(neutrophil extracellular traps,NETs,该机制作用下的中性粒细胞死亡被称为NETosis)在胞外捕获病原菌,并结合胞内分泌的抗菌肽和水解酶等物质对病原菌进行杀灭[66]。GSDM 一方面可以调控细胞焦亡与中性粒细胞死亡,进而破坏致病菌扩繁的微环境以及促进NETosis 过程对病原菌的杀伤;另一方面GSDM 已被发现能够对细菌起到直接杀灭或抑制生长的作用[43-44,67-69]。
尽管GSDM 活化以及膜孔洞的形成在生理病理过程中发挥了关键的作用,但过度的活化会导致炎症相关的疾病,因此其活性需受到严格调控。研究发现静息状态下GSDMD 维持低表达水平,在受到外界刺激后干扰素转录调控因子2 诱导了GSDMD的表达,一定程度上保证细胞中GSDM 足量有效发挥作用[70]。除了转录水平,GSDM 在翻译后也受到调节,活化的凋亡相关的caspase-3 能够介导GSDMD 在靠近N 末端的第88 位天冬氨酸残基后发生剪切,使GSDMD 完全丧失成孔活性,抑制其触发的焦亡过程[4]。有研究发现GSDME 中α1 螺旋的第6 位苏氨酸残基的磷酸化可能通过改变α1 螺旋构象来干扰寡聚化,从而抑制焦亡[64]。另外,活跃的细胞膜修复系统参与了GSDM 孔洞对细胞质膜损伤的调控。研究显示在GSDMD 膜孔洞形成后,钙离子内流引起胞内浓度的瞬间升高由此激活运输所需的内体分类复合体(endosomal sorting complex required for transport,ESCRT)膜修复机制,ESCRT 相关蛋白被招募至膜损伤区域,将损伤部分转移至微囊泡中,并将破损区域以小泡的形式排出胞外,实现膜的修复,缓解GSDMD 孔洞对细胞膜造成的损伤[71]。除ESCRT 之外,其他膜修复机制还包括对损伤膜结构的内吞与利用来自胞内囊泡结构的膜(主要是溶酶体)来修补损伤区域[72-73],这些修复机制是否参与调节GSDM 膜孔洞引发的膜损伤仍待深入研究。
GSDM 家族蛋白可与多种磷脂结合,因此对不同磷脂的偏好性也是影响GSDM 成孔发挥活性的因素。例如鞘磷脂的存在能够促进GSDMD-NT 靶向结合溶酶体[48,74]。在一些刺激因素下,中性粒细胞的GSDMD-NT 会更倾向于结合细胞中的初级颗粒或微管相关蛋白 1A/1B 轻链 3 阳性的自噬体而非细胞质膜[75]。此外,GSDMB 还能结合脑硫脂,但其生理意义仍然未知[76]。
3 Gasdermin 家族蛋白在病理中的作用
3.1 感染性疾病
GSDM 家族,尤其是GSDMD 在机体抗感染过程中发挥了至关重要的作用,其抗感染作用包括直接靶向杀伤或抑制病原菌,以及启动宿主细胞的焦亡程序,上调机体的炎症与免疫应答水平来清除病原菌。多项研究发现GSDMD 的缺失能导致小鼠对弗朗西斯菌(Francisella novicida)、类鼻疽伯克霍尔德菌(Burkholderia pseudomallei)等病原菌的易感性增加[67-77]。然而,GSDM 在感染中并不总是具有对机体的保护作用,其过度激活,一方面会导致免疫细胞的剧烈焦亡和大量耗竭,减弱了机体在感染中后期清除病原体的能力,导致病原体的增殖失去控制,另一方面炎性细胞因子的大量释放又会引发强烈的炎症风暴和器官功能障碍。脓毒症即是临床上一种典型的焦亡程序过度激活所导致的系统性炎症反应综合征,其发病率和致死率较高,病情发展迅速,是人类健康的一大威胁[78]。弥散性血管内凝血(disseminated intravascular coagulation,DIC)是感染引起的另一种过度免疫反应,会导致血液栓塞、休克和脏器功能受损,GSDMD 介导的焦亡可引起细胞膜内侧的磷脂酰丝氨酸发生外翻,进而激活组织因子来诱发外源性凝血[79];依赖于GSDMD活性的中性粒细胞死亡NETosis 过程也会因捕获血小板而导致凝血[80]。在一些经典的小鼠脓毒症和DIC 模型中,Gsdmd敲除小鼠的器官损伤、存活率以及与DIC 相关指标相比野生型小鼠都会显著减轻[2,81]。在病毒感染过程中,来自病毒的核酸和编码蛋白也可被宿主细胞识别并由此触发GSDMD 依赖的细胞死亡,参与宿主的抗病毒免疫反应。在轮状病毒感染过程中,病毒来源的RNA 引起肠上皮细胞中的NLRP9b 炎症小体的组装以及随后GSDMD 依赖的焦亡和IL-18 的成熟释放,小鼠感染模型证实NLRP9b炎症小体组分的缺失可导致小鼠对病毒的易感性增加[82]。两项来自同一实验室的研究分别发现甲型流感病毒和单纯疱疹病毒1 能够引起巨噬细胞的一种焦亡-凋亡-坏死混合死亡形式,并命名为PANoptosis,细胞实验证明GSDMD 参与了该死亡过程[83-84]。GSDMD 的激活对发病程度也可起到促进作用。在鼻病毒和诺如病毒感染中,NLRP3 和GSDMD 依赖性焦亡的发生促进了病理性炎症水平,使得病情加重[85-86]。GSDM 在宿主应对寄生虫感染中也发挥了作用:弓形虫(Toxoplasma gondii)感染小胶质细胞可诱导GSDMD 依赖的细胞因子IL-1α 的释放,小鼠的弓形虫脑部感染模型也表明GSDMD 和caspase-1/11 缺陷小鼠对寄生虫的抵抗能力明显下降[87]。
3.2 肿瘤
众多相关研究显示,GSDM 深度参与了肿瘤与机体的互作,不同的GSDM 在肿瘤发生发展以及机体抗肿瘤免疫中表现出了多样的功能。GSDMA在人胃癌细胞中的表达受到抑制,且其表达能抑制胃癌细胞增殖[21,26]。GSDMB与癌症的关系则更加复杂,根据不同的研究结果分析,GSDMB的功能和表达水平因癌症类型而异:其在宫颈癌、肝癌、结肠癌和人类表皮生长因子受体2 阳性(human epidermal growth factor receptor 2 positive,HER2+)乳腺癌中高表达[88-89],功能性实验发现对GSDMB进行过表达或利用抗体中和GSDMB 蛋白分别促进和抑制了HER2+乳腺癌细胞的转移和侵袭[90-91];与此同时,GSDMB在原发性食管和胃肿瘤中的表达却受到抑制,暗示其对这类癌症可能起到抑癌作用[7]。GSDMC与GSDMB有着类似的作用特点。有研究发现GSDMC在结直肠癌和肺腺癌中高表达,敲除GSDMC可降低结直肠癌细胞的增殖[92-93]。然而,另一项研究表明GSDMC在许多食管鳞状细胞癌中的表达受到抑制[26]。这些研究发现说明GSDMB与GSDMC的作用可能具有肿瘤类型的特异性。GSDME对不同肿瘤具有较为普遍的抑制作用,在各种癌细胞中,GSDME产生了多种突变,其启动子也常发生甲基化的表观遗传修饰,这导致了GSDME表达抑制以及蛋白焦亡活性的丧失[5-6,94-97]。体外实验证明诱导GSDME/Gsdme表达可对胃癌和黑色素瘤的集落形成与肿瘤细胞增殖产生抑制,并降低乳腺癌的侵袭力[64,94-95]。此外,由于GSDME 可在凋亡通路激活时被caspase-3 剪切产生活性NT,且肿瘤组织中经常出现由供氧或营养不足引起的凋亡过程,所以凋亡相关的小分子激动剂也可有效作用于肿瘤细胞。例如用Kirsten 大鼠肉瘤病毒癌基因同源物、表皮生长因子受体或间变性淋巴瘤激酶抑制剂处理肺癌细胞可导致caspase-3 依赖的GSDME 激活,促进了对癌细胞的杀伤[98];另一种凋亡诱导剂依托泊苷对黑色素瘤细胞的毒性效果则会因GSDME的敲除而降低[99]。近期来自Lieberman 实验室的研究发现肿瘤细胞GSDME 的一种新颖活化机制:自然杀伤细胞和白细胞分化抗原8 阳性T 细胞所分泌的颗粒酶B 到达肿瘤细胞内部后可直接切割GSDME并产生与caspase-3 剪切后相同的NT,引起肿瘤细胞的焦亡,促进机体抗肿瘤免疫反应[6]。另外,GSDMD 介导的细胞焦亡也能够促进抗肿瘤进程。丝氨酸二肽酶8/9 的小分子抑制剂Val boro Pro 可激活人类髓系细胞中的caspase-募集结构域成员8 炎症小体,诱导caspase-1 和GSDMD 依赖的焦亡发生。该效应特异性地作用于大多数人急性髓系白血病(acute myeloid leukemia,AML)细胞系和原发性AML 细胞,引发肿瘤细胞的死亡[100]。
3.3 炎症相关疾病
由于GSDM 家族尤其是GSDMD 在炎症中起到重要作用,GSDM 和上游相关通路的异常活化会引起自发炎症,导致非感染相关的免疫性疾病。家族性地中海热(familial mediterranean fever,FMF)是一种自发性常染色体隐性遗传病,由地中海热基因(mediterranean fever gene,MEFV,编码Pyrin 蛋白)的突变引起,该突变降低了Pyrin 炎症小体对激活信号的感受阈值,使得Pyrin 变得易于活化,该病通常表现为炎性疾病综合征,包括反复发热和腹膜炎。GSDMD 作为Pyrin 下游的关键效应蛋白对FMF 的病理至关重要。在Pyrin 自激活的小鼠FMF模型中,敲除Gsdmd可有效抑制疾病小鼠的炎症症状[101]。新生儿发病的多系统炎症性疾病(neonatal onset multisystem inflammatory disease, NOMID) 也存在类似的病理机制,这是一种罕见的自身炎症性疾病,由炎症小体NLRP3 编码基因的突变导致的自激活引起,发生于婴儿期,症状为发热、慢性脑膜炎、皮疹和关节病等。研究表明抑制NOMID 模型小鼠体内的GSDMD 活性也可有效减轻其症状[14]。多基因突变引起的疾病例如多发性硬化(multiple sclerosis,MS)也依赖GSDMD 的活性。在模拟人类MS 的小鼠实验性自身免疫性脑脊髓炎模型中可检测到GSDMD 的激活,且Gsdmd的敲除或针对GSDMD 的抑制剂处理可有效下调小鼠的神经损伤水平[13,102]。此外,GSDMD 还参与调控了器官或组织移植中的机体损伤,糖尿病及其并发症,创伤性脑损伤以及一些由非感染损伤导致的疾病,且大都对病理过程起到了促进作用[103-112]。相关研究通过小鼠实验证明其中一些疾病的病理表型可以被GSDMD的表达下调或GSDMD 蛋白活性抑制所减轻,但GSDMD 在更多疾病中的贡献和作用机制仍有待于进一步研究。
GSDM 家族其他成员与非感染性疾病的发生也存在联系,例如人GSDMA基因与系统性硬化病、炎症性肠病和儿童哮喘的发生相关[113-116]。小鼠Gsdma3的不同突变则与皮肤表型有关,包括角膜溃疡和脱毛等[23,117-121]。GSDMB的表达水平或单核苷酸突变率与哮喘、克罗恩病、1 型糖尿病和溃疡性结肠炎的易感性成正相关[16,113,116,122-127]。GSDME和PJVK,则都被发现与听力障碍有关,二者的突变分别造成常染色体显性和常染色体隐性的遗传性听力损伤[17,40,128-129]。
4 Gasdermin 家族蛋白作为多种疾病潜在的治疗靶标
由于GSDM 家族在感染、炎症和肿瘤相关疾病中具有重要地位,以其作为靶点进行药物和疗法的研发已成为一个新兴的方向。在炎症反应中,GSDMD 是多种炎症小体下游共同的效应蛋白,且其成孔效应同时调控了细胞的死亡和炎性细胞因子的释放,可以说GSDMD 是控制炎症反应的关键,因此控制GSDMD 的激活可能是治疗感染或非感染性炎症疾病的有效策略:抑制GSDMD 的过度激活,理论上能够有效降低炎症水平,防止病原感染引起炎症风暴和器官损伤,或减轻自身免疫性疾病患者的症状;诱导GSDMD 激活或基因表达的上调则可能有助于放大炎症反应,在一些情况下(例如疫苗注射时)增强机体免疫应答能力。不过鉴于GSDM 家族在炎症中的节点地位,对GSDM 的干预可能会对机体免疫功能造成深远的影响,因此相关治疗策略需经过仔细的临床检验。GSDM 家族蛋白在癌症中则具有更加复杂的作用,不同的GSDM在不同类别的癌症中分别显示出促癌或抑癌效果,相关研究还存在许多空白。另外,由于GSDM 家族在正常组织和免疫细胞中也广泛存在,激活或抑制GSDM 来治疗癌症可能会导致较大的副作用 ,例如有研究尝试用诱导GSDME 激活的方式抑制AML 癌细胞生长,但同时却导致了广泛的组织损伤,并且由于诱导剂非特异性激活了免疫细胞中的GSDMD 导致免疫细胞焦亡[5]。综合来看,以GSDM 作为癌症治疗的靶点需进一步广泛深入地研究并谨慎对待。
迄今为止,已有多种能够直接靶向并调控GSDM 活性的小分子化合物被发现。一种被美国食品药品监督管理局批准已用于临床的戒酒药——双硫仑(disulfiram,DSF)近期被证明能通过修饰GSDMD 的第191 位半胱氨酸残基来抑制其成孔活性[102],不同的动物模型实验证明DSF 对GSDMD介导的炎症性疾病具有显著保护作用[13,102]。具有类似机制的小分子抑制剂还包括坏死磺酰胺和富马酸二甲酯,二者都可通过靶向GSDMD 半胱氨酸残基的方式抑制其活性,在小鼠相关疾病模型中表现保护作用[102,130]。还有不同研究报道化合物LDC7559和punicalagin 可抑制GSDMD 依赖的细胞死亡过程,但详细机制仍有待解析[53,131]。
5 总结和展望
焦亡现象初次发现于上世纪90 年代,其伴随着巨噬细胞对细菌侵染的响应而发生,这种现象曾被视为细胞凋亡。由于该细胞死亡形式依赖于caspase-1 的活性,并伴随着促炎症因子的释放,因此在2001 年Cookson 等人将其命名为“pyroptosis”(细胞焦亡)。近年来,对这一新兴细胞死亡形式的机制研究得到迅速发展。2015 年GSDM 家族蛋白GSDMD 作为细胞焦亡的关键执行分子被发现使得焦亡的范畴得到了进一步扩展,其相关信号网络的图景也逐步完善。相关研究也显示了GSDM 家族在感染、炎症相关疾病和癌症中的重要作用。基于GSDM 的治疗策略也在逐渐提出和确立。时至今日,细胞焦亡仍属于免疫学研究的热点方向。
尽管如此,焦亡领域仍然有大量的问题等待解答。GSDM 激活信号强弱的胞内调控机制仍然不明;GSDM 家族中仍存在一些成员,例如GSDMA和DFNB59,其激活机制、生理功能缺乏深入研究;GSDM 家族在各类疾病中所扮演的角色也亟待进一步揭示。相关研究会将GSDM 及其介导的焦亡过程同更多的生理和病理过程联系起来,理清GSDM 相关信号途径在疾病中的作用脉络,并将这些基础研究拓展至临床应用,例如开发更多靶向GSDM 的疾病疗法,寻找更具特异性的GSDM 抑制剂/激动剂和更多活性调控途径,同时建立GSDM 活性和相关病理指标的关联性,提高相关疗法的精确性,减少副作用,以期GSDM 和细胞焦亡领域的研究为人类健康事业做出更多贡献。
