运用生物信息学方法筛选肾透明细胞癌中差异表达的关键基因
2022-10-19陆凌翔蒋民军陈建春陆季成
陆凌翔, 蒋民军, 陈建春, 陆季成
(江苏省苏州市第九人民医院, 1.泌尿外科, 2.肿瘤科, 江苏 苏州, 215000)
全世界每年有超过40万人罹患肾细胞癌(RCC), 发病年龄大多为60岁左右,其中2/3患者为男性[1]。RCC包括多种亚型,约70%为肾透明细胞癌(ccRCC)[2]。早期确诊并尽早治疗可以提高ccRCC的治愈率,但多达1/3的ccRCC患者确诊时已出现转移[3]。转移性ccRCC恶性程度高,严重威胁患者的生命健康[4],因此寻找其诊断和治疗靶点尤为重要。微小RNA(miRNA)是一类长度为19~23 nt的内源性小型非编码类RNA,由DNA转录而来[5]。miRNA可通过直接与靶向信使RNA(mRNA)上的碱基进行配对,引导RNA诱导沉默复合体(RISC)间接降解所编码的蛋白质,或通过直接抑制mRNA蛋白的翻译而在转录后水平或亚转录阶段终止后水平间接调节靶蛋白质的表达[6]。许多特殊的miRNA在ccRCC组织和正常肾组织中存在差异表达,如微小RNA-206(miR-206)、微小RNA-141-3p(miR-141-3p)、微小RNA-30a(miR-30a)和微小RNA-194-5p(miR-194-5p)[7-10]。研究[11]发现,微小RNA-182-5p(miR-182-5p)通过靶向泛素结合酶E2T mRNA抑制泛素结合酶E2T蛋白表达,进而抑制ccRCC细胞的增殖、迁移和侵袭。另有研究[12]发现,近80%的与ccRCC转移相关的miRNA与ccRCC患者的复发率和生存率有关。RNA干扰可有效控制基因的表达,在肿瘤治疗中起着广泛作用[13], 而miRNA与ccRCC的发生及发展密切相关,故调控相关miRNA的表达有望成为治疗ccRCC的新方法。本研究对数据库样本进行挖掘和分析,筛选ccRCC组织的差异表达基因(DEGs), 旨在寻找ccRCC治疗的潜在生物学靶点,现报告如下。
1 材料和方法
1.1 数据来源
从基因表达综合(GEO)数据库(https://www.ncbi.nlm.nih.gov/geo/)的数据集中下载ccRCC样本和正常肾组织样本的表达谱数据。① GSE116251数据集中,从18名ccRCC患者(9名疾病复发和9名未复发患者)的配对肿瘤组织和邻近正常组织中分离总RNA,然后进行NanoString miRNA分析。NanoString是一种高通量的RNA表达检测方法,其优点是适用于严重降解的RNA样本如福尔马林固定石蜡包埋(FFPE)样本,且操作简单、数据准确、重现性好。应用实时定量聚合酶链反应(PCR)方法对肿瘤中失调的miRNA 进行进一步验证。② GSE168845数据集中,分析4个ccRCC组织和配对癌旁组织样本(作为对照)的基因表达谱。
1.2 方法
从GEO数据库(https://www.ncbi.nlm.nih.gov/geo/)的GSE116251、GSE168845数据集中下载ccRCC样本和正常肾组织样本表达谱数据,利用R软件的Limma包筛选出差异表达的miRNA和基因,设置|log2FC|≥2和错误发现率(FDR)<0.05为筛选标准。FunRich(http://www.funrich.org/)是一个主要用于基因和蛋白质的功能富集和相互作用网络分析的独立软件工具,还可用于靶基因预测。本研究利用FunRich网站对差异表达miRNA(DEmiRNAs)进行潜在转录因子及靶基因预测,并进行生物学过程(BP)、细胞成分特征(CC)和分子功能(MF)富集分析。利用R软件的ClusterProfiler包和Cytoscape软件对靶基因的信号通路进行富集分析。将预测到的靶基因与DEGs取交集,获得差异表达的靶基因,通过Cytoscape软件绘制miRNA-mRNA调控网络。以DEmiRNAs及其靶基因的中位表达值为分界,将患者分为高表达组和低表达组,通过Kaplan-Meier plotter网站(https://kmplot.com)判断DEmiRNAs及其靶基因的表达水平与患者总生存期的关联,选择P值最小的靶基因及其相互作用的miRNA与肿瘤基因组图谱(TCGA)中616例ccRCC患者的临床特征(年龄、性别、TNM分期和初始治疗结局)进一步分析。通过人类蛋白质图谱(HPA)数据库验证所选基因在ccRCC组织及正常组织中的蛋白表达。
1.3 统计学分析
使用Wilcoxon检验筛选肿瘤组织与癌旁组织的DEmiRNAs、DEGs, 采用BH法对P值进行矫正,设置|log2FC|>2和FDR<0.05为筛选标准。采用卡方检验评估各指标在高表达组、低表达组中的分布差异。采用Kaplan-Meier分析和Log-rank检验评估高风险组、低风险组患者的生存差异。应用R软件(版本4.0.2)对数据进行统计学分析,P<0.05为差异有统计学意义。
2 结 果
2.1 DEmiRNAs和DEGs的筛选
应用R软件筛选GSE116251数据集中的DEmiRNAs和GSE168845数据集中的DEGs,分别筛选出3种DEmiRNAs(miR-142-3p、miR-200c-3p、miR-141-3p)和1 610种DEGs, 见图1。
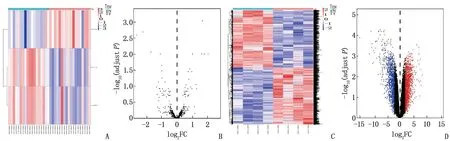
A: GSE116251中DEmiRNAs的热图; B: GSE116251中DEmiRNAs的火山图; C: GSE168845中DEGs的热图;D: GSE168845中DEGs的火山图。红色代表上调,蓝色或绿色代表下调。
2.2 基因本体论(GO)和京都基因和基因组百科全书(KEGG)富集分析
GO和KEGG富集分析结果显示, DEmiRNAs在细胞环腺苷酸(cAMP)受体介导的信号传导、翻译的调节、发展、溶酶体、高尔基体、细胞表面、受体信号蛋白和丝氨酸酶/苏氨酸肌激酶活性、磷酸二酯水解酶活性和细胞骨架蛋白等方面具有独特的功能,见图2; DEmiRNAs主要富集于6种途径,即破骨细胞分化、金黄色葡萄球菌感染、细胞黏附分子、补体和凝血级联、吞噬体和原发性免疫缺陷,见图3。
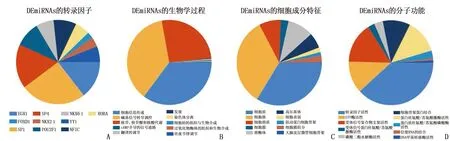
A: FunRich软件识别的DEmiRNAs的潜在转录因子; B、C、D: miRNA靶基因表达的前10种生物学过程、细胞成分特征和分子功能富集分析。
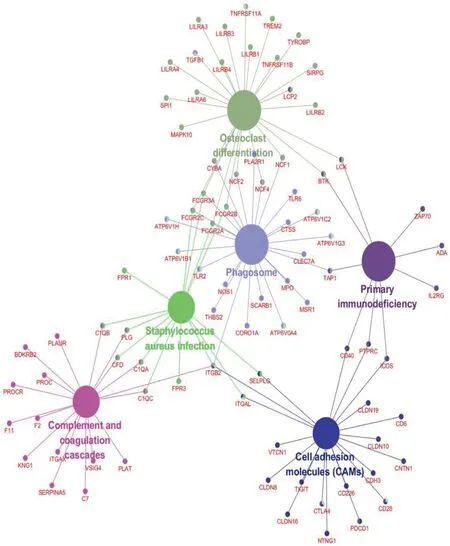
Osteoclast differentiation: 破骨细胞分化; Phagosome: 吞噬体;Primary immunodeficiency: 原发性免疫缺陷;Staphylococcus aureus infection: 金黄色葡萄球菌感染;complement and coagulation cascades: 补体和凝血级联;Cell adhesion molecules(CAMs): 细胞黏附分子。
2.3 miRNA-mRNA调控网络
使用预测软件对3个DEmiRNAs的靶基因进行预测,共获得846个靶基因,将其与GSE168845数据集的DEGs取交集,共得到49个交集靶基因,见图4A。根据相互作用关系,筛选出miRNA-mRNA调控网络, miRNA包括hsa-miR-142-3p(上调)、hsa-miR-200c-3p(下调)和hsa-miR-141-3p(下调),其关联的30个靶基因中包括11个下调基因和19个上调基因,见图4B。
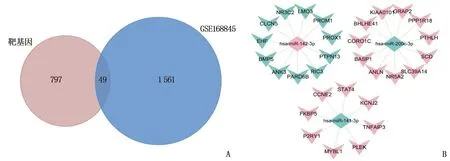
A: 预测的靶基因与GSE168845数据集DEGs交集的韦恩图; B: miRNA-mRNA调控网络(绿色表示下调,粉色表示上调)。
2.4 miRNA、基因表达水平与ccRCC患者总生存期的关系
通过Kaplan-Meier plotter网站分析ccRCC患者的总生存期,结果显示, miR-142-3p、miR-200c-3p、miR-141-3p和TNFAIP3、STAT4、P2RY1、CORO1C、MYBL1、CCNE2、BHLHE41、ANLN、BASP1均与ccRCC患者总生存期相关(P<0.05), 见图5、图6。各种基因中,ANLN基因的P值最小即差异最显著,故本研究选择ANLN基因及其相互作用的miR-200c-3p进行后续研究。
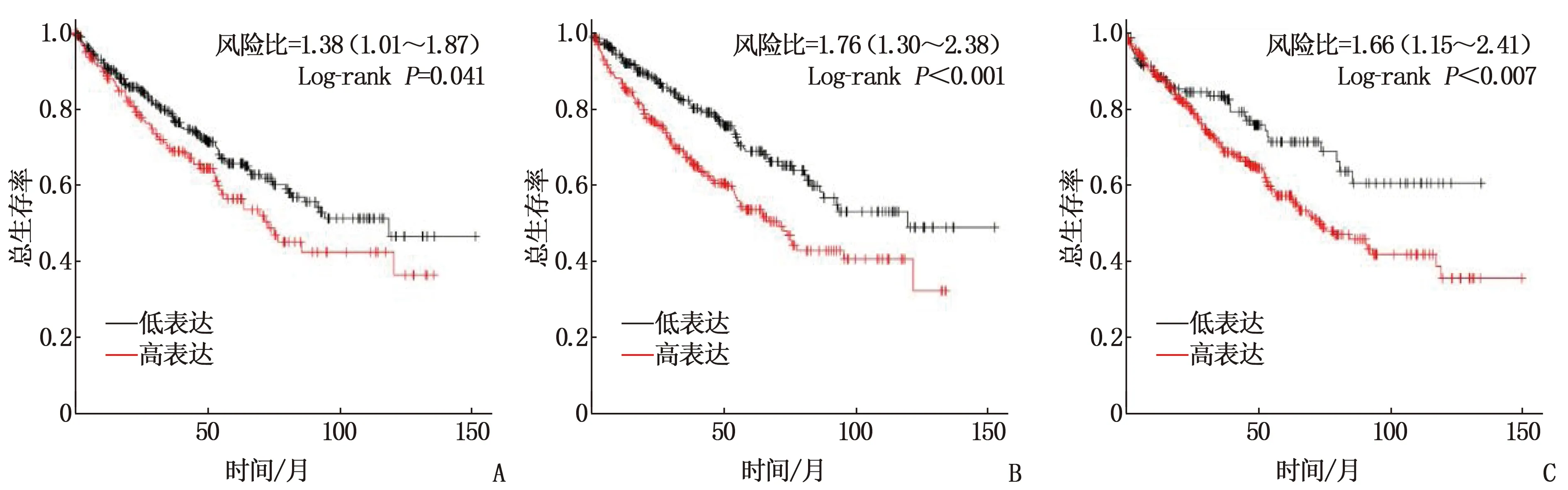
A: hsa-miR-141-3p与总生存期的关系; B: hsa-miR-142-3p与总生存期的关系; C: hsa-miR-200c-3p与总生存期的关系。
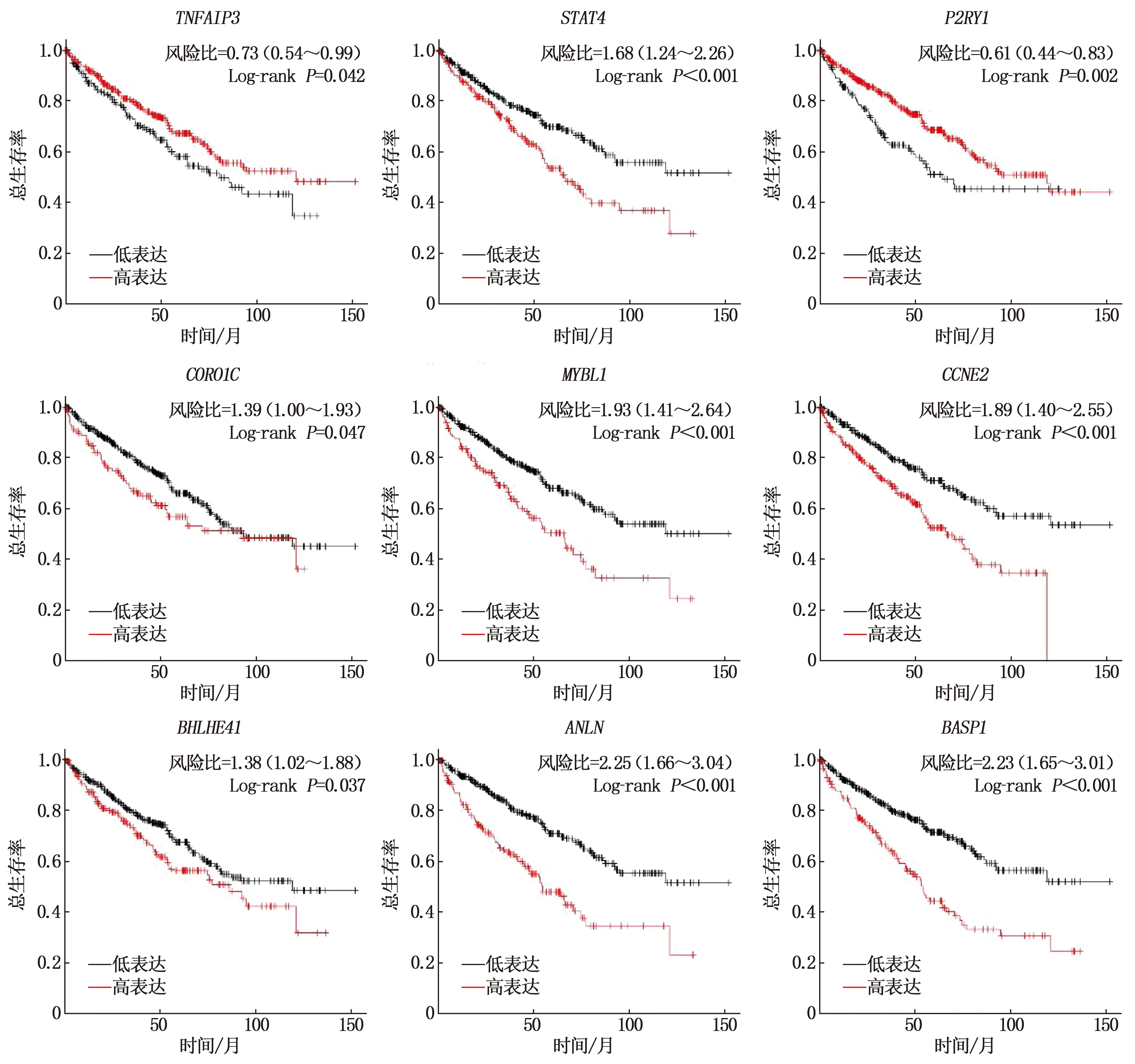
图6 不同基因表达水平与ccRCC患者总生存期的关系
2.5 ccRCC的临床特征和mRNA表达
从TCGA数据库中下载ccRCC患者临床诊断指标和相关基因片段的克隆表达研究数据(例如患者的年龄、性别、TNM分期和初始治疗结局数据),结果显示,不同TNM分期患者的ANLN、miR-200c-3p表达水平比较,差异有统计学意义(P<0.05), 见表1、表2。进一步分析ccRCC患者临床特征与预后的关系后发现, TNM分期、初始治疗结局均与ccRCC患者预后相关(P<0.05), 见表3、表4。

表1 ANLN表达水平与ccRCC患者临床特征的关系
表2 miR-200c-3p表达水平与ccRCC患者临床特征的关系
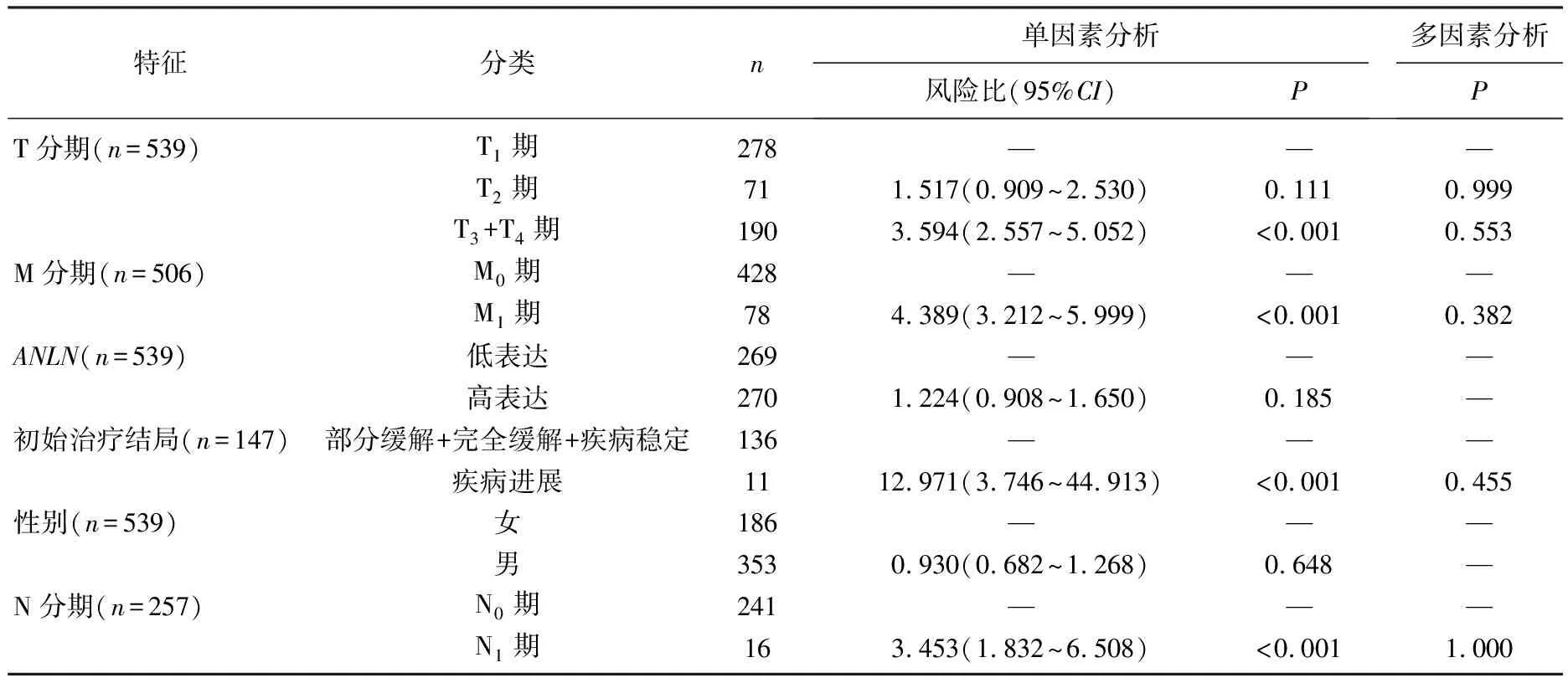
表3 ccRCC患者临床特征、ANLN表达与预后关系的单因素分析和多因素分析

表4 ccRCC患者临床特征、miR-200c-3p与预后关系的单因素分析和多因素分析
2.6 免疫组织化学(IHC)方法验证ANLN表达
基于HPA数据库,本研究发现,与正常肾脏组织相比, ccRCC组织中的ANLN表达显著上调,见图7。
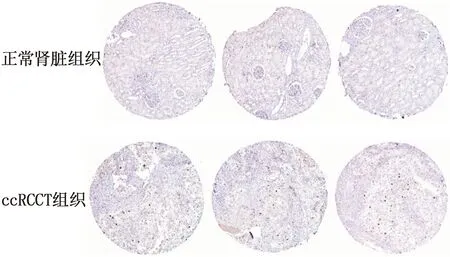
图7 ANLN蛋白在正常肾脏组织(n=3)和ccRCC组织(n=3)中的表达(HE染色,放大100倍)
3 讨 论
本研究从GEO数据库下载GSE116251、GSE168845数据集进行分析, KEGG分析结果显示, DEmiRNAs主要富集于破骨细胞分化、金黄色葡萄球菌感染、细胞黏附分子、补体和促凝血因子级联反应、吞噬体和原发性免疫缺陷这6个途径中,而这些途径与癌症的发生和转移密切相关[14-16]。已有研究[17]表明,细胞黏附分子配体唾液酸Lewis(a/x)抗原可通过与E-选择素受体结合而参与内皮细胞壁的黏附,这一生化过程由炎症细胞和肿瘤细胞共同参与,可以部分解释炎症与肿瘤发生之间的关系,进一步阐明抗炎药在肿瘤治疗中的功效。转录调节因子是一种能够以特定序列结合DNA分子并参与调节基因转录活动的蛋白质[18]。转录抑制因子可通过选择性识别一些特定类型的DNA序列信息来同时调控染色质形成和转录,从而形成一个可调控整个基因组信息表达活动的复杂系统。此外,转录因子可以结合特定序列促进或抑制下游基因,对肿瘤发生、迁移和侵袭等生物学过程产生重要影响[19]。
本研究使用Cytoscape软件进行miRNA-mRNA调控网络构建,确定了3个miRNA(miR-142-3p、miR-200c-3p和miR-141-3p), 并获得了846个潜在靶基因,其中仅49个在GSE168845数据集中差异表达,进一步根据相互作用关系筛选miRNA-mRNA调控网络, 3个miRNA关联的30个靶基因中包括11个下调基因和19个上调基因。miR-142-3p是杂合性丢失、易位和扩增的首选位点,可参与人体内多种特定生物类型细胞上的某些原发细胞肿瘤分子中过氧化酶的表达,包括原发性肝细胞癌、卵巢癌和胰腺癌等[20-23]。此外, miR-142-3p的过表达可以抑制血清剥夺诱导的细胞凋亡,当miR-142-3p抑制剂降低miR-142-3p表达时,可以逆转对细胞凋亡的抑制,且miR-142-3p可在G1/S期加速细胞分化并促进细胞增殖[24]。包含miR-200a、miR-200b、miR-200c、miR-141和miR-429的miR-200家族已被证明在上皮间质转化中发挥作用[25]。另外,越来越多的研究[26-27]表明miR-200家族成员在多种恶性肿瘤组织中下调,包括RCC。因此, miR-200c是一种潜在的生物标志物,或可为RCC替代治疗方案的制订提供参考依据。
ANLN基因编码蛋白中含有的Anillin蛋白,是体内一种有着高度保守性表达的肌动蛋白结合蛋白,在胞质的分裂及其复制形成过程中发挥着关键作用[28]。研究[29-31]发现, ANLN在许多原发肿瘤组织中均有表达,特别是晚期胰腺癌、乳腺癌和中晚期肺癌组织。流行病学研究[32-33]发现,ANLN还具有一系列生物功能,包括参与调节人类肿瘤干细胞的生长、增殖、转化和肿瘤迁移,影响多种恶性肿瘤组织的发生与发展,因此其通常被认为是对某些肿瘤患者预后具有重要意义的几个主要特征基因之一。高ANLN表达水平的原发性乳腺癌患者预后可能较差,而ANLN水平敲低可能导致乳腺癌组织衰老的细胞增加、多核形态和G2/M期停滞[34]。另一项人类非小细胞肺癌细胞凋亡实验[35]发现,敲除ANLN会显著抑制肿瘤细胞分裂增殖速度和增加多形核细胞凋亡数量。
近年来, miRNA及其靶基因在肿瘤发生发展中的作用已被广泛研究,基因表达调控可能成为肿瘤治疗的新选择。本研究发现, miR-200c-3p及其靶基因ANLN参与了ccRCC的发展,并可能成为ccRCC的潜在生物标志物。ANLN可能对ccRCC患者的预后判断具有重要价值,并可为其治疗策略提供新的选择。
