唑啉草酯原药含量测定方法研究
2022-10-15段文胜刘益红
段文胜,刘益红
(泸州东方农化有限公司,四川 泸州 646300)
唑啉草酯是先正达成功开发的选择性除草剂,主要用于牙后防除小麦和大麦田一年生禾本科杂草,其活性高,起效快,对作物安全,对环境友好,自2006年上市以来,市场已遍布全球多个国家,逐渐成为除草剂家族的大品种[1]。有关唑啉草酯含量测定方法有液相色谱法[2-3],有资料显示唑啉草酯分解受pH值影响较大[4],针对检测过程中试样稳定性问题未见有文献报道。本文采用高效液相色谱法将唑啉草酯及其中杂质分离,并考察了影响检测试样稳定性的因素,采用外标法准确测定唑啉草酯的含量。该方法分离效果理想,重复性好,准确度高,结果满意。
1 试验部分
1.1 仪器及试剂 LC—20ADVP高效液相色谱仪;SPD—M20A二极管阵列检测器;LabSolution色谱数据工作站。唑啉草酯标准品99.0%;甲醇 色谱纯;乙腈 色谱纯;磷酸 优级纯;超纯水。
1.2 色谱条件 色谱柱:SinoChrom ODS-BP5μ 250mm×4.6mmid;流动相:甲醇/水(75/25) (每500mL流动相中含0.25g磷酸);流速:1.0mL/min;检测波长:UV257nm;柱温:40℃;进样量:5μL。
1.3 试验方法
1.3.1 唑啉草酯标准溶液配制 准确称取唑啉草酯标准品约20mg(精确至0.01mg)于50mL容量瓶中,加乙腈/水(体积比5/5,含0.1%磷酸,下同)溶解,并稀释至刻度,摇匀,备用。
1.3.2 样品溶液的配制 准确称取唑啉草酯原药样品约20mg(精确至0.01mg)于50mL容量瓶中,加乙腈/水溶解,稀释至刻度,摇匀。
1.3.3 样品的测定 在选定的色谱条件下,分别吸取标准(样品)溶液5μL,进样,重复操作,保证其相对标准偏差<0.5%,记录唑啉草酯的峰面积,并取其平均值。样品中唑啉草酯的质量分数X按下式计算。
式中:
A1—样品中唑啉草酯峰面积平均值;
A2—标样中唑啉草酯峰面积平均值;
m1—称取样品的质量,mg;
m2—称取标样的质量,mg;
P—唑啉草酯对照品的质量分数,%。
2 结果与讨论
2.1 色谱条件的选择
2.1.1 色谱柱的选择 采用通用型反相色谱柱分别对样品进行分离,本文对比了Hedera ODS-2柱、SinoChrom ODS-BP柱、Diamonsil C18柱不同生产厂家的3种色谱柱,结果发现在同样的流动相条件下,无明显区别,本文选用柱压较低的SinoChrom ODS-BP柱。
2.1.2 流动相的选择 采用甲醇(或乙腈)/水(或磷酸盐缓冲液等)体系分别对唑啉草酯样品进行色谱分离,发现有机相采用乙腈唑啉草酯分离不理想,分离度差,峰形不对称,采用甲醇可获得较好的分离度和峰形,考虑到酸性体系有利于抑制唑啉草酯分解,在流动相中加入适量磷酸可实现检测过程稳定。所以本文选用甲醇/水做流动相,并对其配比及磷酸加入量进行了调整,最后确定为甲醇/水(75/25)(每500mL流动相中含0.25g磷酸),其色谱图(图1)。

1-溶剂;2-唑啉草酯分解杂质;3,4,6-未知杂质;5-唑啉草酯图1 唑啉草酯液相色谱图(乙腈做有机相配样)
2.1.3 特异性及检测波长的选择 采用二极管阵列检测器分别对唑啉草酯标样及样品溶液进行光谱数据采集,获得唑啉草酯标样及样品的紫外波长扫描图(图2、图3)。从图中可以看到唑啉草酯标样及样品分别在202、257nm有较大吸收峰,且202nm较257nm强,但考虑到低波长基线干扰较大,所以本文选用257nm作为检测波长。

图2 唑啉草酯标样紫外吸收谱图
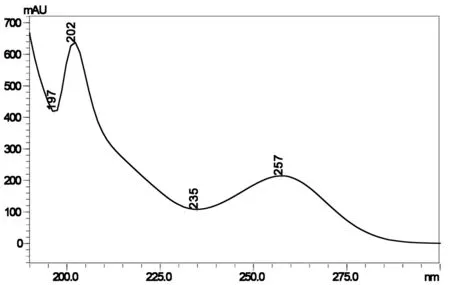
图3 唑啉草酯样品紫外吸收谱图
2.2 配样溶液选择 在分析条件选择过程中发现试样会缓慢分解成2#峰杂质(图1、图4),且使用甲醇做有机相配制样品,会生成4#峰杂质(图4)。针对试样不稳定情况对乙腈体系配样溶液进行了选择,准确称取同一样品分别用乙腈、乙腈/水(5/5,含0.1%磷酸,pH值约为2.6)、乙腈/水(5/5,含0.2%磷酸,pH值约为2.3)配样,在实验规定的条件下连续进样10次,计算唑啉草酯峰面积与分解杂质峰面积比,以进样次数为横坐标,峰面积比为纵坐标做图(图5),由图可以发现乙腈/水酸性溶液配制的试样稳定性较好,考虑到酸性太强会影响色谱柱寿命,所以本文选择乙腈/水(5/5,含0.1%磷酸)做为配样溶液。
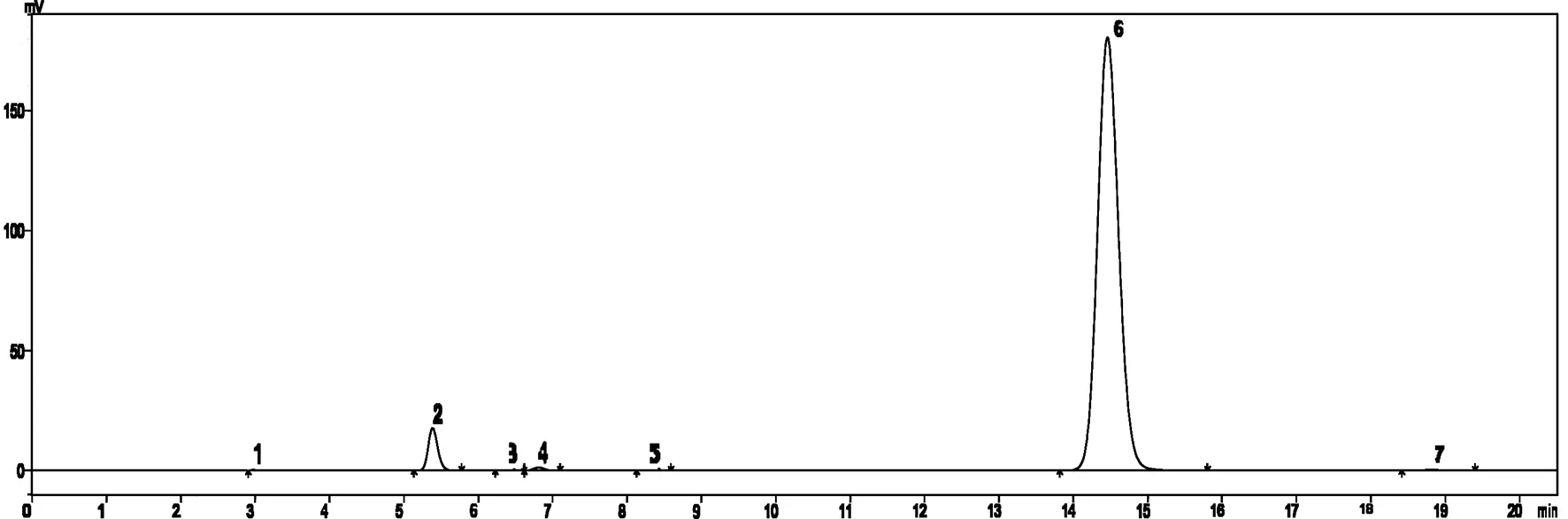
1-溶剂;2-唑啉草酯分解杂质;3,5,7-未知杂质;4-唑啉草酯甲醇溶解产生杂质;6-唑啉草酯图4 唑啉草酯液相色谱图(甲醇做有机相配样)
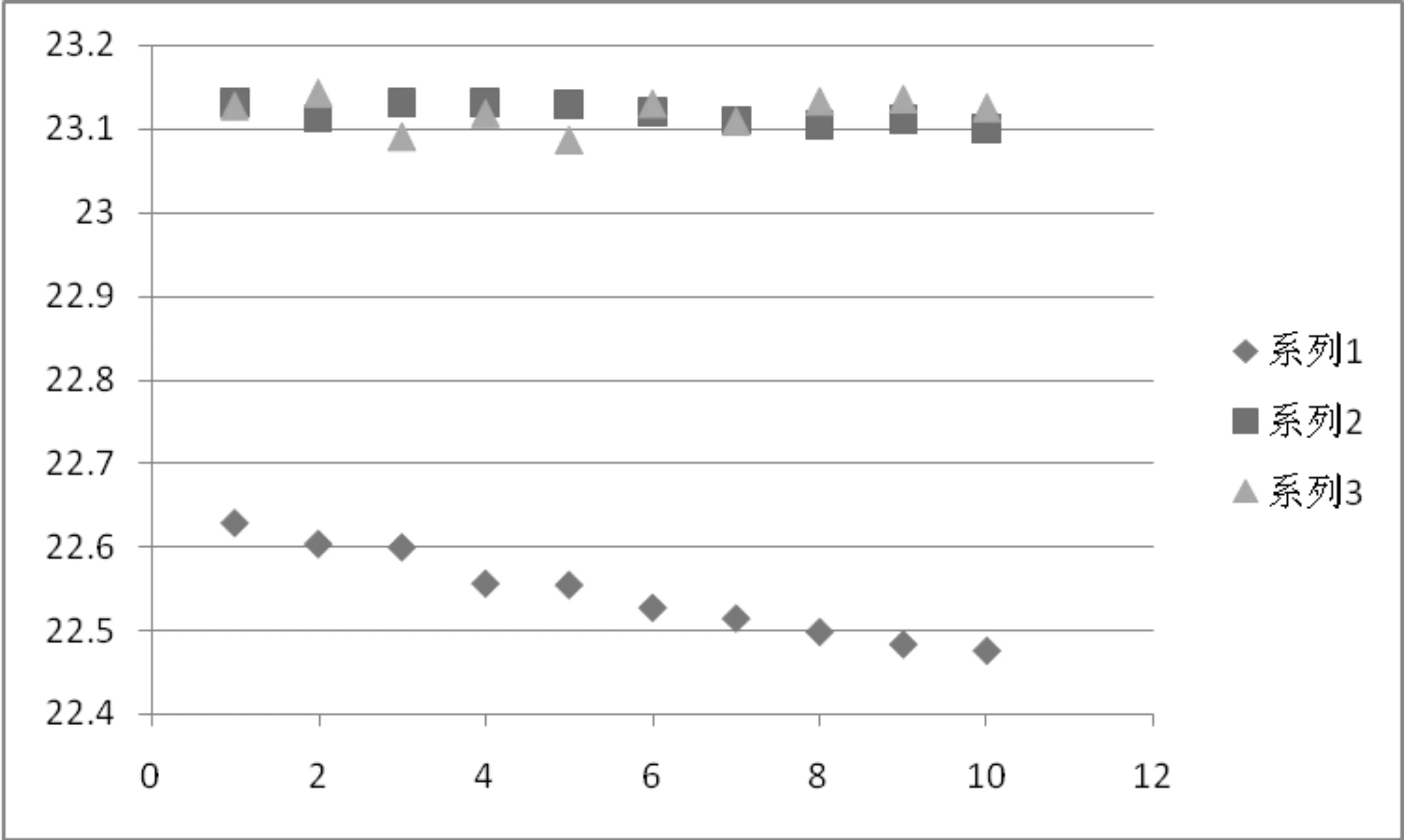
系列1-纯乙腈配样(称样量:19.64mg);系列2-乙腈/水(0.1%磷酸)配样(称样量:20.46mg);系列3-乙腈/水(0.2%磷酸)配样(称样量:20.57mg);图5 唑啉草酯试样稳定性
2.3 分析方法的线性相关性试验 准确称取唑啉草酯标准品150mg(精确至0.01mg)于50mL容量瓶中,加乙腈/水溶解,并稀释至刻度,摇匀(标准贮备液)。分别准确吸取0、2、4、6、8、10mL,分别加入到6个50mL的容量瓶中,加乙腈/水稀释至刻度,摇匀。根据试验规定 的操作方法测定,记录唑啉草酯色谱峰面积,以唑啉草酯面积与浓度作图,测得唑啉草酯标准曲线为Y=9 510.41C-14 923.31,r=0.999 8,表明唑啉草酯在0~590.79mg/L的浓度范围内呈线性(表1、图6)。

表1 分析方法的线性相关性试验数据
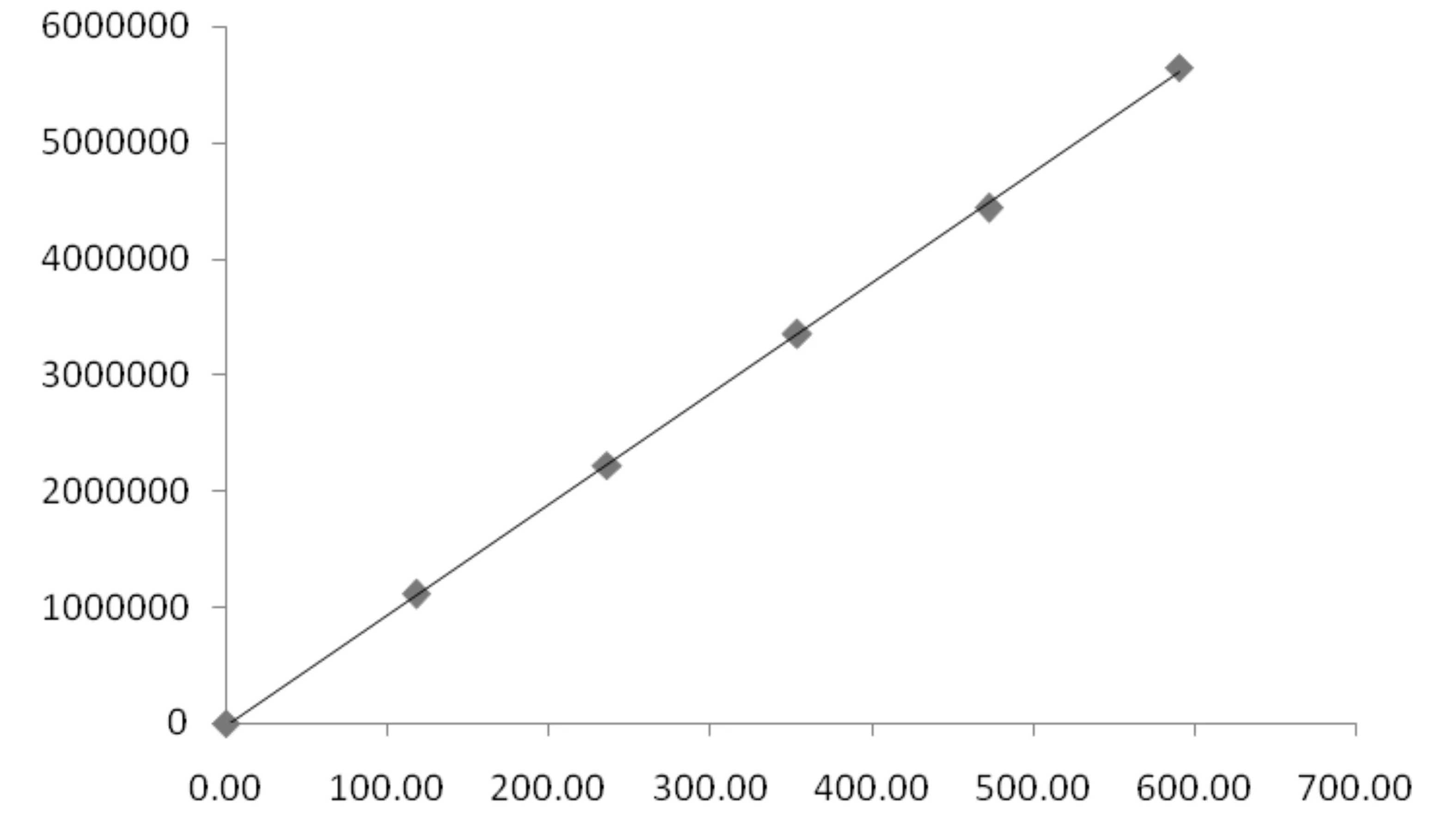
图6 唑啉草酯线性关系图
2.4 分析方法的精密度试验 准确称取同一样品,按试验规定方法,配制6份溶液,在上述实验条件下进行分析,测唑啉草酯的标准偏差为0.26,RSD为0.27%(表2)。

表2 分析方法的精密度试验结果
2.5 分析方法的准确度试验 分别准确称取已知含量的样品溶液约20mg 6份,各分别加入实验2.3配制的唑啉草酯标准贮备液1、2、3mL,按试验规定的方法配成一系列溶液,测定唑啉草酯的总量,并计算回收率(表3)。
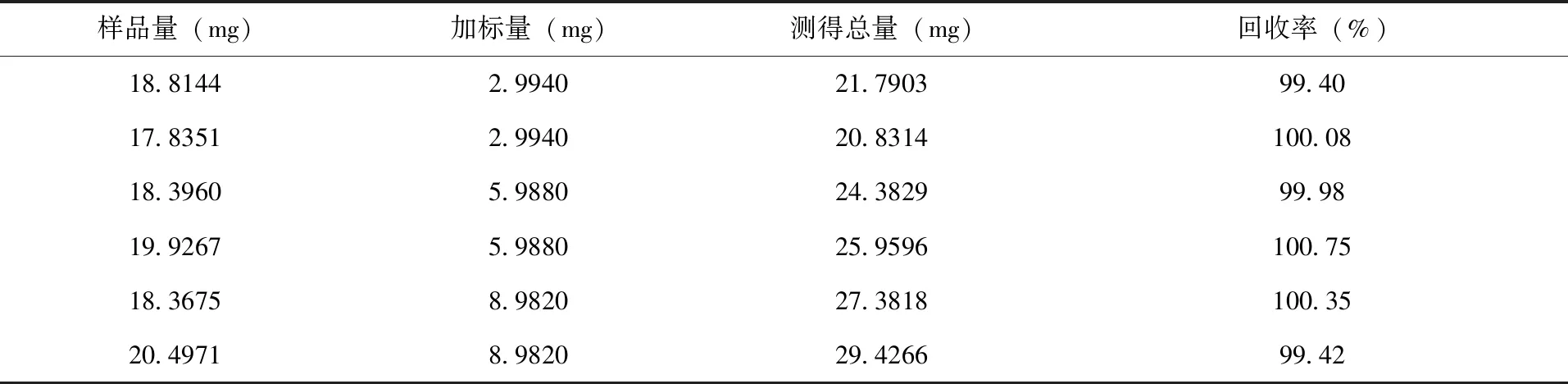
表3 分析方法的准确度试验结果
本文建立的高效液相色谱法测定唑啉草酯含量的方法,可避免唑啉草酯在检测过程中缓慢分解,有效保证了唑啉草酯长时间检测试样稳定,使检测结果更准确,重复性更好,实践证明是一种确实可行的好方法。
