高效液相色谱法测定毒氟磷原药及可湿性粉剂中有效成分含量
2022-10-15佟盟露丁玉玲贾孙悦马腾飞姜宜飞
佟盟露,丁玉玲,贾孙悦,马腾飞,周 芹,3,4*,姜宜飞
(1.黑龙江大学现代农业与生态环境学院,黑龙江 哈尔滨 150080;2.黑龙江大学化学化工与材料学院,黑龙江 哈尔滨 150080;3.农业农村部甜菜品质监督检验测试中心,黑龙江 哈尔滨 150080;4.农业农村部糖料产品质量安全风险评估实验室,黑龙江 哈尔滨 150080;5.农业农村部农药检定所,北京 100125)
引言
毒氟磷是一种新型α-氨基膦酸酯类杀菌剂,化学名称为N-[2-(4-甲基苯并噻唑基)]-2-氨基-2-氟代苯基-O,O-二乙基膦酸酯[1],是一种新型高效的抗病毒制剂,能够激活植物的系统性抗性(SAR)[2]。植物病毒是第二大类植物病害[3],变异速度快,不断出现新毒株[4],具有危害大,难检测以及难防治等特点[5],因此,给农业生产带来了极其严重的危害,为了防止植物病毒病的大规模爆发,贵州大学历经十年的研究,发现了具有全新结构的磷系抗植物病毒新农药-毒氟磷,并获得农业部新农药登记,成为具有完全自主知识产权的农药品种[6]。
毒氟磷具有主动防御,促进生长以及内吸作用等基本特性[7],还可以调节植物的内源生长因子,恢复植物叶部功能及根部生长。对植物病毒也有很高的防治效果[8],其中对黄瓜、烟草以及番茄病毒都有防治作用[9],此外,毒氟磷对水稻黑条矮缩病等的防治效果也非常显著,Li等[10]经实验表明,毒氟磷在体内和田间试验中都能抑制水稻黑条矮缩病毒在水稻中的感染活性,与SRBSDV P9-1基因表达的抑制活性最高,且在体外与SRBSDV P9-1蛋白有微弱的亲和力,最终证实毒氟磷与P9-1八聚体蛋白内孔中的精氨酸175结合而抑制SRBSDV的感染,具有良好的应用前景。
如今,农药的安全性问题被越来越多的人关注。毒氟磷作为一种新型的环保型抗病毒有机磷农药,被广泛普及应用,但对毒氟磷的体内安全性评价非常有限[11],且由于毒氟磷的高对映选择性和高控制成本,导致毒氟磷的非手性分析以及残留物测定仍没得到充分的发展。郑坤明等[12]采用高效液相色谱测定了大棚和露地条件下,西瓜中毒氟磷的残留量在0.01~5.0mg/kg添加水平下,毒氟磷平均回收率分别为84.5%~103.9%,RSD为1.2%~7.5%,方法的精密度和准确度良好。焦斌等[13]建立了土壤中毒氟磷残留的检测方法,采用QuEChERS前处理,应用高效液相色谱串联质谱进行样品检测,在准确度、灵敏度和精密度上均满足残留分析要求,可以为毒氟磷在中国典型土壤中的残留检测提供方法支撑。目前的研究报道多数是关于毒氟磷在植株,土壤及水体中的残留问题,毒氟磷制剂有效成分检测方面的报道比较少,不少企业为了降低农药成本,提高农药防治效果,在产品中添加非法成分[14],隐性添加不易察觉,尚未建立系统的农药成分监管方法[15],陈书华等[16]在市场抽检的生物农药样品中,禁限用农药成分检出率高达42.9%,农药添加未经登记的农药成分已经影响了农药质量安全。因此,对农药制剂有效成分的检测非常必要,本实验拟建立一种方法可以同时适用于毒氟磷原药(technical material简写为TC)及可湿性粉剂(water power简写为WP)中有效成分的测定。采用反相高效液相色谱法,通过波长扫描确定最适宜的检测波长,特异性检验确定峰纯度,优化了流动相比例,建立的方法完全满足上述要求,具有灵敏度高、操作简单的优点,同时为国家标准的制定提供依据。
1 材料与方法
1.1 实验材料
1.1.1 实验仪器 Agilent 1260型高效液相色谱仪(配有二极管阵列检测器);色谱柱为250 mm×4.6mm(i.d.)不锈钢柱,内装 ZORBAX SB-C18、粒径为5μm;针式过滤器(配有孔径为0.45μm的滤膜);50μL微量进样器;5μL定量进样管;超声波清洗器。
1.1.2 实验试剂 色谱纯甲醇;新蒸二次蒸馏水或超纯水;已知质量分数为99.1%的毒氟磷标样。
1.2 实验方法
1.2.1 液相色谱条件 流动相:Ψ(甲醇:水)=80:20,经滤膜过滤,并进行脱气;流速:1.0 mL/min;柱温:35 ℃±2 ℃;检测波长:270 nm;进样体积:5μL。
1.2.2 测定步骤
医院门诊大厅有一块区域是门诊服务中心,有大量自助服务设备,提供建档、预约、挂号、查询、充值等自助服务,涵盖患者就诊全流程。这只是医院自助机总量的一部分,钮罗涌介绍,全院不同角落分布着150多台自助服务机。
1.2.2.1 标准溶液的制备 将0.05g(需要精确到0.000 1g)毒氟磷标样,放入体积为100mL的容量瓶中,加入一定体积的甲醇,使用超声波清洗器进行超声,超声的时间是5~10min,将其冷却到室温,最终用甲醇稀释定容至刻度线,摇匀备用。
1.2.2.2 试样溶液的制备 将含有0.05g(需要精确到0.000 1g)毒氟磷的试样,放入体积为100mL的容量瓶中,加入一定体积的甲醇,使用超声波清洗器进行超声,超声的时间是5~10 min,将其冷却到室温,最终用甲醇稀释定容到刻度线,摇晃均匀,过滤备用。
1.2.2.3 测定 在这个操作条件之下,等到仪器稳定之后,将多针标样溶液连续注入进去,直到毒氟磷相邻2针的峰面积之间的相对变化<1.2%时,按照标样溶液、试样溶液、试样溶液、标样溶液的顺序进行测定。
2 结果与分析
2.1 实验条件的选择
2.1.1 吸收波长的选择 毒氟磷结构中含有共轭双键,在紫外区会有吸收,为了确定毒氟磷最适宜的吸收波长,利用高效液相色谱配制的二极管阵列检测器,对毒氟磷溶液进行扫描,波长范围是190~400nm(图1是毒氟磷的紫外光谱图),图中可见,毒氟磷在227nm处有最大的吸收波长,在270nm处也有较强吸收,用这2个最大吸收波长对毒氟磷进行检测,结果表明,虽然在227nm处灵敏度更高,但是基线的平衡时间过长,而且在227nm处一些杂质会有紫外吸收,因此,为了检测减少其他杂质对毒氟磷的干扰,确定其检测波长为270 nm[17]。
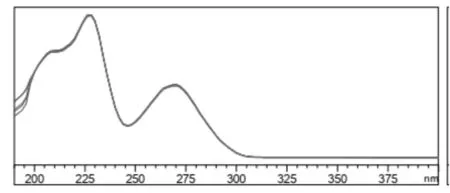
图1 毒氟磷的紫外光谱图
2.1.2 色谱柱及流动相的选择 通常而言,流动相的比例配制以及流速的设置都对复杂样品的分离有着很大的影响,因此,实验前必须要优化流动相配比,本实验选择的色谱柱是反相色谱柱,内装ZORBAX SB-C18。根据毒氟磷物理和化学性质以及溶剂的紫外吸收波长,用甲醇来溶解样品,流动相用甲醇和水,对不同比例的甲醇/水体系进行比较,选择最适的流动相比例。结果发现,随着甲醇比例的增加,保留时间逐渐减小,出峰时间过早,会有一些杂质干扰;而如果水含量过高,保留时间过长,峰形较宽,拖尾,而且灵敏度也会随之降低,因此,最终选择的流动相是Ψ(甲醇:水)=80 :20,以1.0mL/min为流速。在这个条件下,能够呈现出较好的峰形,而且能够彻底与其他物质分开,精密度和准确度良好,并且在10min之内就可以达到基线分离,分析时间较短,提高了工作效率。在优化的流动相比例及检测波长条件下,毒氟磷出峰时间大约在8.1min(图2~图4毒氟磷标准品、原药及可湿性粉剂的液相色谱图)。
2.2 峰纯度检验 一个分析方法建立后,需要对其专属性进行验证[18],峰纯度(Peak purity)检查越来越多的被用于评价方法专属性,可以提供峰纯度的检测器有二极管阵列检测器(DAD)、质谱(MS)及多通道紫外可见光检测器等等。峰纯度成为是否能够准确分析的重要因素[19]。本试验采用HPLC-DAD峰纯度分析法对毒氟磷进行鉴别,30%毒氟磷可湿性粉剂以及98%毒氟磷原药中的毒氟磷的峰纯度因子全部都>990,有效成分处没有被其他杂质所干扰,符合标准(图5~图6)。

保留时间/min图2 毒氟磷标准品的的液相色谱图
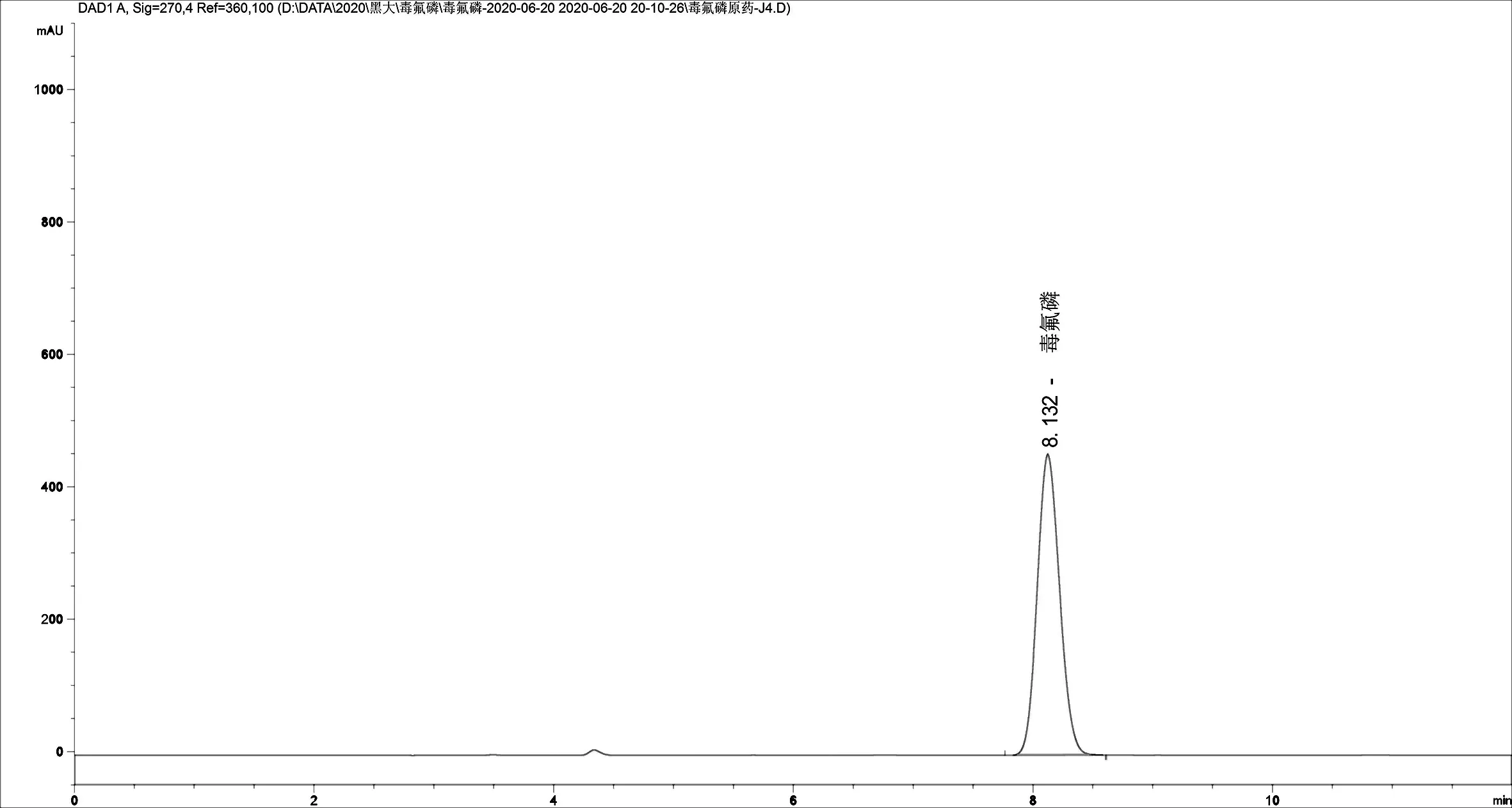
保留时间/min图3 毒氟磷原药液相色谱图
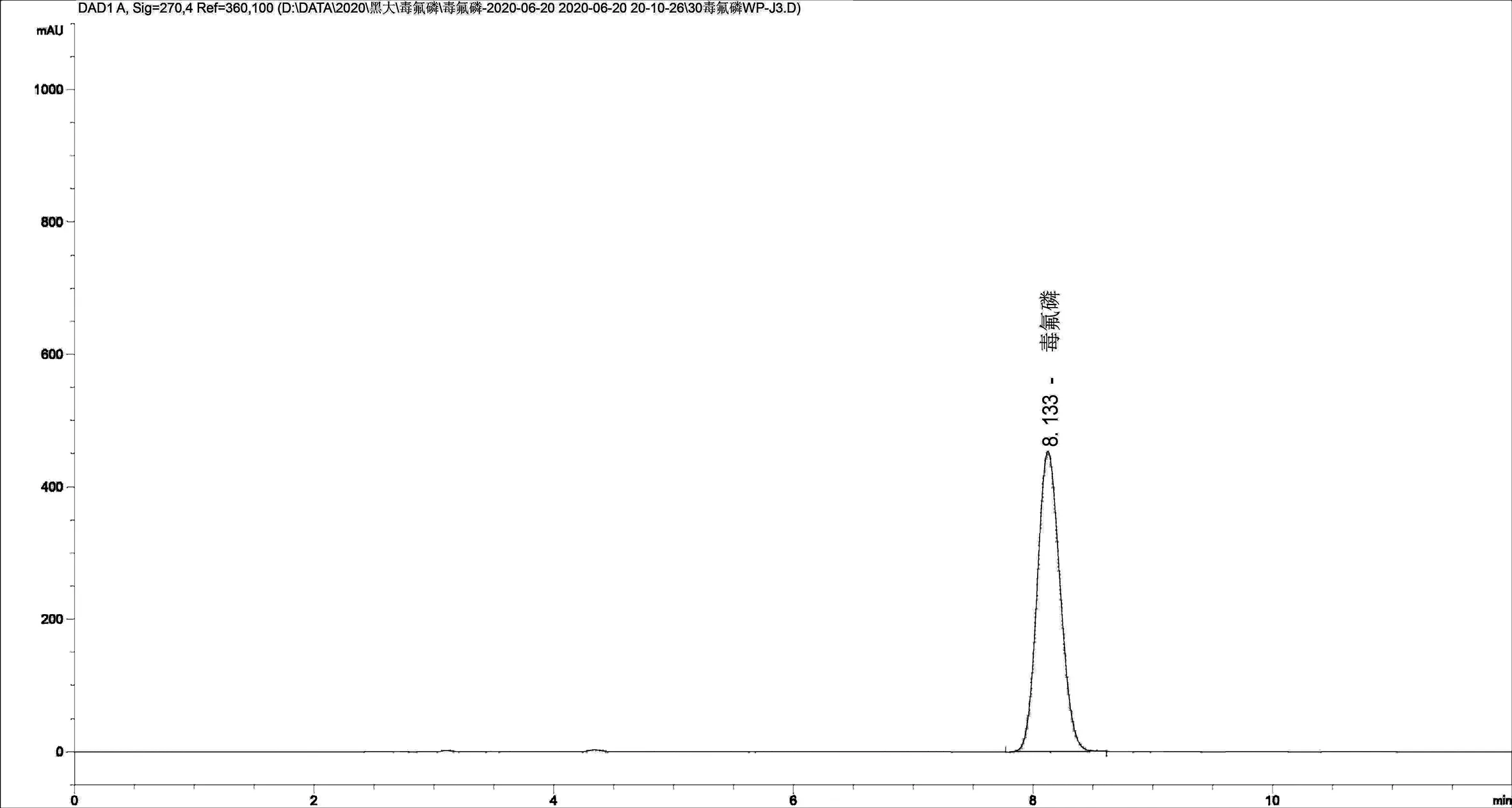
保留时间/min图4 毒氟磷可湿性粉剂液相色谱图
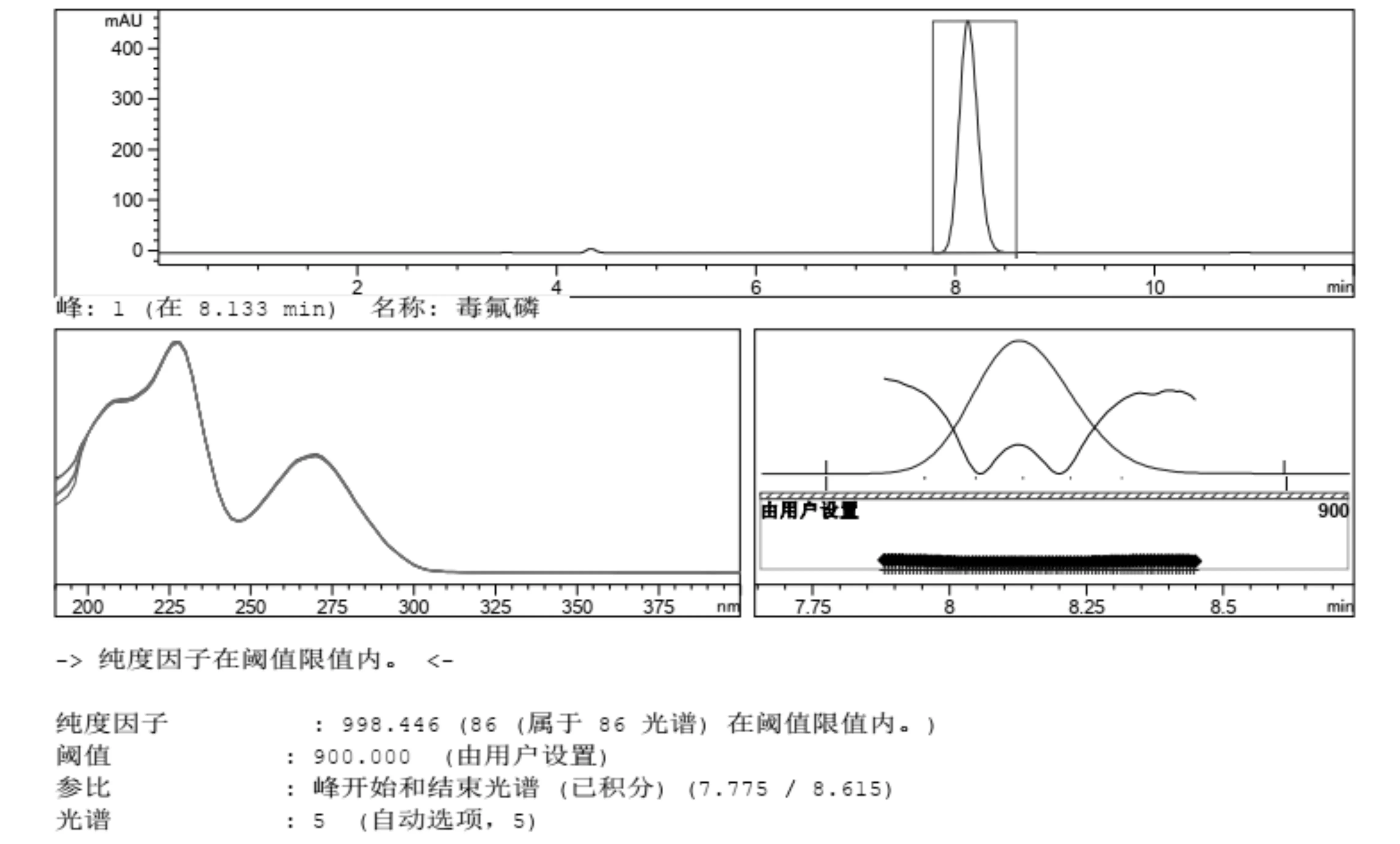
图5 98%毒氟磷原药HPLC-DAD峰纯度色谱图
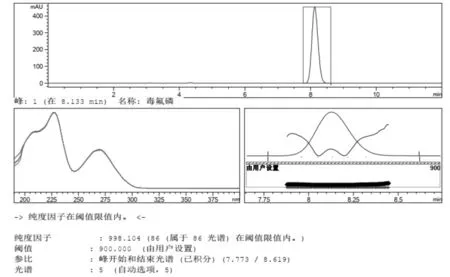
图6 30%毒氟磷可湿性粉剂HPLC-DAD峰纯度色谱图
2.3 方法学考察

图7 毒氟磷峰面积与质量浓度关系图
2.3.2方法精密度试验 精密度为同一标准样品多次测定的变异系数[20],通常用偏差,标准偏差以及相对标准偏差来表示。本实验对同一样品进行5次测定,计算结果的标准偏差以及相对标准偏差。按照1.2.2试样溶液的制备方法,分别配制5个98%毒氟磷原药精密度溶液,标记为TC-A1至TC-A5。5个30%毒氟磷可湿性粉剂精密度溶液,从WP-A1到WP-A5依次进行标记。将STD3作为标液,按照以上的操作条件,待仪器基线稳定后,在标样溶液、精密度溶液、精密度溶液、标样溶液的顺序下,依次进行测定,结果(表1、表2)。
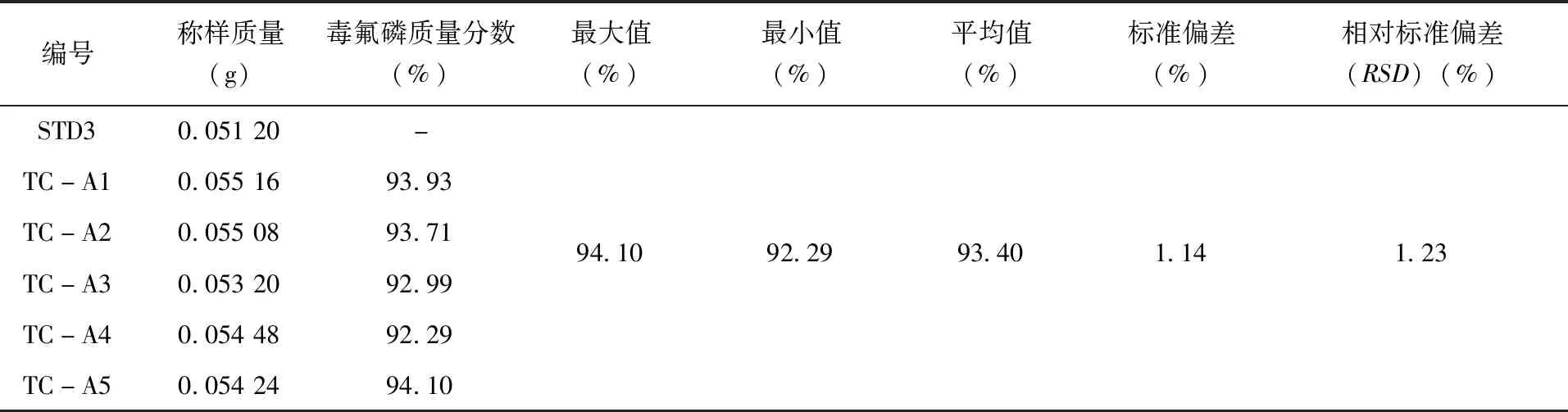
表1 98%毒氟磷原药中毒氟磷精密度试验结果
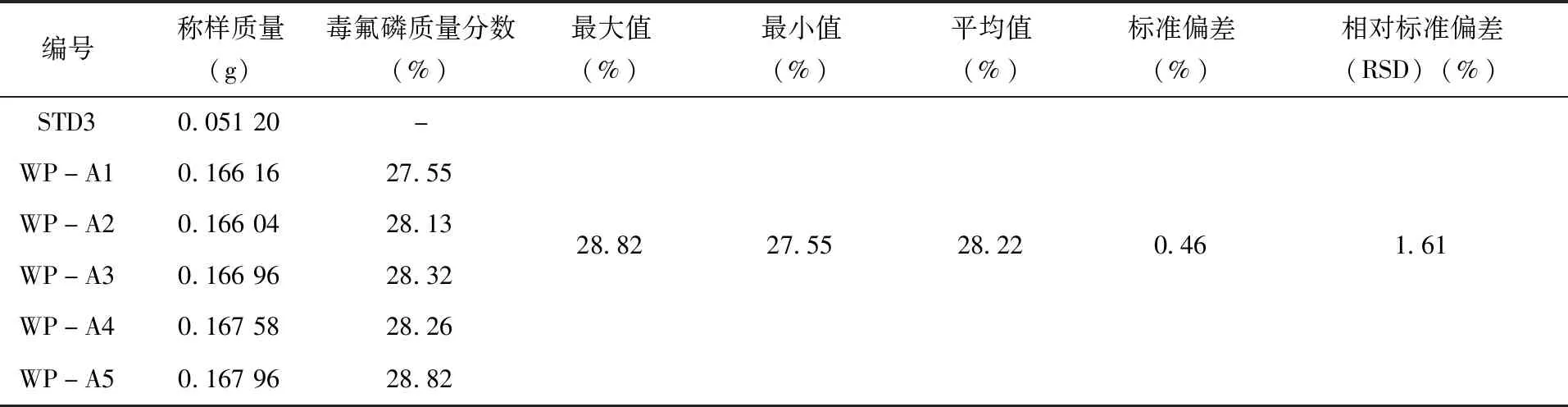
表2 30%毒氟磷可湿性粉剂中毒氟磷精密度试验结果
实验结果表明,98%毒氟磷原药中毒氟磷质量分数测定结果的RSD为1.23%,小于修改的Horwitz公式2(1-0.51ogC)×0.67=1.35(C=0.934 0),30%毒氟磷可湿性粉剂中毒氟磷质量分数测定结果的RSD为1.61%,小于修改的Horwitz公式2(1-0.51ogC)×0.67=1.62,(其中C=0.282 2),测定出的精密度符合标准。
2.3.3 方法准确度试验 称取含0.025g(精确至0.000 1g)毒氟磷的30%毒氟磷可湿性粉剂于放入体积为100mL容量瓶中,再加入0.025g(精确至0.000 1g)的毒氟磷标样,同1.2.2的方法对溶液进行配置,选择5个有效成分准确度溶液,从WP-B1到WP-B5依次进行标记。将线性相关溶液STD3作为标样溶液,待仪器基线稳定后,在标样溶液、准确度溶液、准确度溶液、标样溶液的顺序下,依次进行测定,结果(表3)。实验结果表明,30%毒氟磷可湿性粉剂中毒氟磷平均回收率为99.43%,具有良好的准确度。
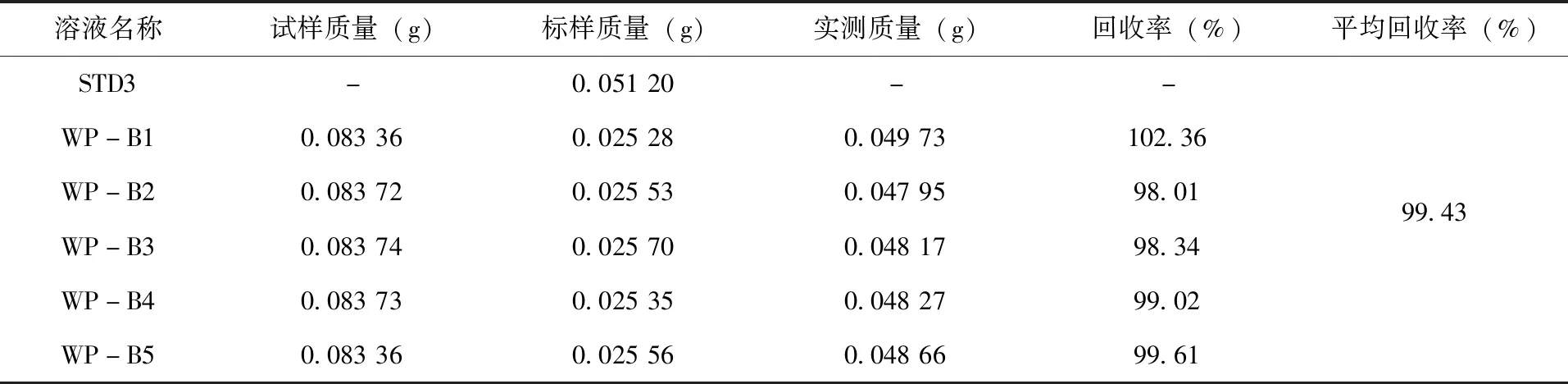
表3 30%毒氟磷可湿性粉剂回收率试验结果
2.3.4 不同实验室方法验证 为了验证所建立的能够同时适用于98%毒氟磷原药和30%毒氟磷可湿性粉剂中有效成分检测方法能够适用于每一个不同的实验室,因此按照已经建立好的方法,对4个不同的实验室进行协同验证试验。每组4家单位在2个不同日期对样品含量进行重复测定,分别计算试样1和试样2的结果,每个样品2个不同日期测定得到4个结果,(表4、表5)。

表4 98%毒氟磷原药协同验证试验结果
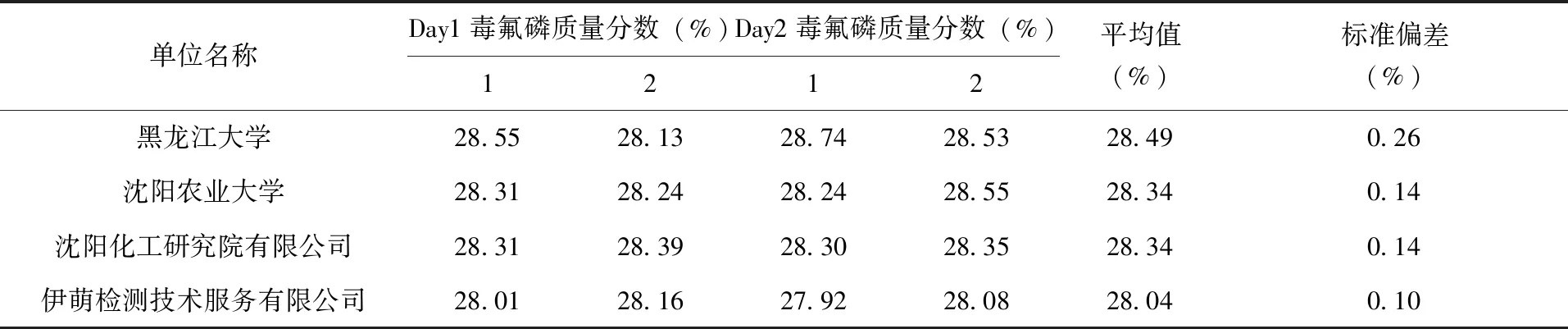
表5 30%毒氟磷可湿性粉剂协同验证试验结果
结果表明,98%毒氟磷原药、30%毒氟磷可湿性粉剂的重复性相对标准偏差RSDr分别为0.562 7%、0.557 3%,再现性相对标准偏差RSDR分别为0.733 9%、0.816 2%,都小于相应的Horwitz公式2(1-0.51ogC)理论计算值,表明不同单位间的检测结果符合性良好,所建立的毒氟磷液相色谱分析方法能够解决日常检测工作的需求,结果(表6)。

表6 不同实验室方法验证结果统计表
3 讨论
高效液相色谱拥有诸多优点,方便快捷,具有较高的准确性,可以检测到微量成分,因此被广泛使用,然而目前的研究结果主要集中于毒氟磷残留的检测方面,而对于农药制剂有效成分等方面的研究内容相对较少,且多数研究结果都只建立了测定一种试剂有效成分的高效液相色谱分析方法,如葛家成等[21]建立了一种测定氟氯虫双酰胺悬浮剂有效成分的高效液相色谱分析方法,王睿等[22]建立一种对40%环丙唑醇悬浮剂进行分离和测定的高效液相色谱分析方法,而本研究能够同时测定98%毒氟磷原药、30%毒氟磷可湿性粉剂中有效成分含量的高效液相色谱分析方法。
本试验还按照已经建立好的方法,对4个不同的实验室进行协同验证试验,数据表明,不同单位间的检测结果符合性良好,充分满足日常检测工作的需要。
4 结论
本研究建立的毒氟磷测定方法,98%毒氟磷原药、30%毒氟磷可湿性粉剂相对标准偏差(RSD)分别为1.23%和1.61%;30%毒氟磷可湿性粉剂的回收率为99.43%,不同实验室不同仪器之间的重复性标准偏差(RSDr)分别为0.562 7%和0.557 3%,再现性相对标准偏差(RSDR)分别为0.733 9%、0.816 2%。该方法能够获得较高的精密度和准确度,且线性关系良好,能够满足毒氟磷含量分析的要求,操作简单,快捷,为98%毒氟磷原药、30%毒氟磷可湿性粉剂质量控制提供了一种可行的方法。
