《已上市化学药品药学变更研究技术指导原则(试行)》原料药生产工艺变更的解读
2022-10-11王淑华
王淑华
国家药品监督管理局药品审评中心
杨文智
国家药品监督管理局药品审评中心
许真玉*
国家药品监督管理局药品审评中心
《已上市化学药品药学变更研究技术指导原则(试行)》[1]为《药品注册管理办法》[2]和《药品管理法》[3]的配套文件,变更原料药生产工艺是《已上市化学药品药学变更研究技术指导原则(试行)》的一个重要的章节,本文结合指导原则起草的思路,从变更原料药的合成路线、生产工艺及工艺参数、起始原料、起始原料/中间体内控标准及生产过程控制、生产设备等5 个方面对如何进行变更分类进行归纳总结,并对各项变更分类的研究工作进行列表对比,同时说明了需要重点关注的事项。
1 变更分类的界定
变更原料药生产工艺主要包括变更原料药的合成路线、生产工艺及工艺参数、起始原料、起始原料/中间体内控标准及中间过程控制、生产设备等5 种变更情形。本文以列表方式对原料药生产工艺的变更分类进行解读,详见表1。

表1 原料药生产工艺的变更分类
建议关注以下几点内容:①变更越靠近成品风险越高,特别是变更最后一步及之后的生产条件,风险较高。如变更结晶溶剂的种类、重结晶步骤增加活性炭处理、变更最后一步反应的溶剂等均属于重大变更。②变更合成路线的,变更后起始原料的选择应符合ICH Q11 的要求,建议结合ICH Q11 的相关要求,充分评估起始原料选择的合理性。③对于多数批次都要进行的返工,建议应当作为一个工艺步骤纳入注册生产工艺中,属中等变更。④增加重新加工工艺属重大变更,需要进行全面的研究验证。⑤增加新的生产过程控制方法或制订更严格的过程控制限度,一般属于微小变更,但是如果上述变更是由于原料药生产过程中发现存在工艺缺陷或稳定性问题而进行的,则应按照重大变更进行申报。⑥所有导致原料药关键质量属性变化的生产工艺变更均属于重大变更。
2 研究验证工作
变更原料药生产工艺需要进行的研究验证工作,详见表2。
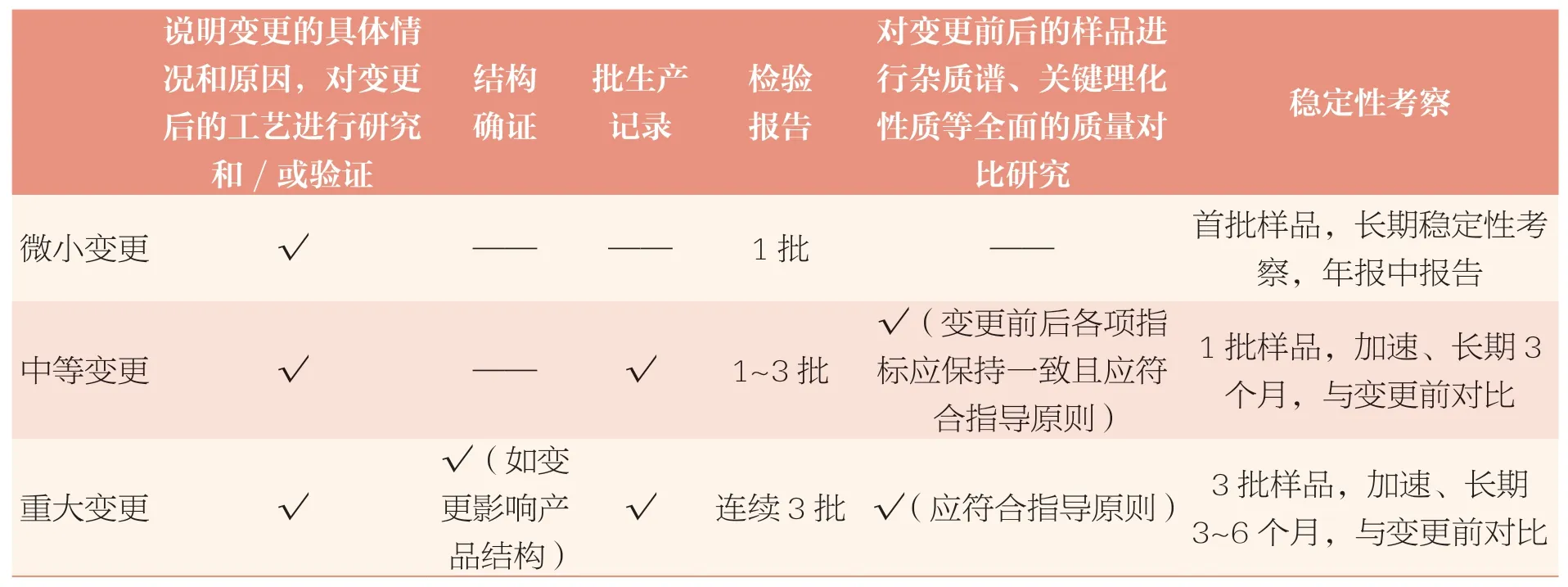
表2 变更原料药生产工艺需要进行的研究验证工作
建议关注以下几点内容:①建议采用商业化生产规模样品进行研究,如采用中试规模样品,需提供充分的理由。②重点关注杂质谱、关键理化性质的变化。③参考ICH M7,关注潜在基因毒性杂质的研究。④如变更前后样品的质量不一致,例如杂质谱、理化性质发生重大变化,应按照重大变更申报补充申请。⑤部分变更可能需要增加稳定性研究批次和考察时间,如某些不稳定的药物。部分变更在充分评估的基础上,可能不需要针对变更进行稳定性研究,如变更某些中间体的内控标准。⑥提供稳定性研究资料的同时,应承诺按照稳定性研究方案考察长期稳定性并在年报中进行报告。⑦当原料药发生变更时,应及时通知制剂持有人,制剂持有人对相应制剂进行评估和研究。
当对比研究结果符合以下条件时,可认为变更前后杂质谱一致:①新增杂质未高于《化学药物杂质研究的技术指导原则》及ICH Q3A 等规定的鉴定限度;②已有杂质(包含立体异构体)及杂质总量均在质量标准规定的限度内,如标准中无规定,应在原工艺生产的多批产品测定范围内;③新使用的溶剂残留量符合《化学药物残留溶剂研究的技术指导原则》及ICH Q3C 等的有关规定;④新的无机杂质符合《化学药物杂质研究的技术指导原则》及ICH Q3D 等的有关要求。⑤应参考ICH M7 对致突变杂质进行考察,必要时进行控制。
3 结语
本文结合《已上市化学药品药学变更研究技术指导原则(试行)》从另外一个维度,通过变更分类的界定、研究验证工作两个方面对原料药的生产工艺变更进行了解读,同时说明了需要重点关注的事项,以帮助相关单位更好地理解指导原则的内容。同时,以期能为相关单位进行变更研究及监管机构进行监管提供参考和借鉴。
