Ru纳米团簇/MoO3-x纳米带双功能催化剂的合成及其肼辅助全解水性能
2022-10-10常亚楠陆徐云马张玉许冬冬包建春
常亚楠 陆徐云 马张玉 许冬冬 刘 影 包建春
(南京师范大学化学与材料科学学院,南京210023)
0 引言
氢气(H2)由于其清洁和可再生等优势,被认为是化石燃料的最佳替代品之一[1-3]。与传统的制氢方法相比,电催化全解水(OWS)具有环境友好、操作简便和制氢纯度高等优点。OWS由阴极氢析出反应(HER)和阳极氧析出反应(OER)两个半反应组成[4-6]。然而,该方法中OER过程涉及多电子转移步骤(4OH-→O2+2H2O+4e-,1.23 V(vs RHE)),不仅降低了制氢效率,还消耗了大量能量,被认为是OWS技术发展的瓶颈[7-9]。虽然已经设计和合成了一些电催化剂,但仍需要高于1.5 V的电压驱动OWS。用热力学/动力学上更有利的小分子氧化反应替代阳极OER可以降低OWS的工作电压,具有良好的发展前景。基于此,人们用甲醇[10]、甲酸[11]、尿素[12]、肼[13]和乙二醇[14]等物质进行了探索。在这些分子中,肼氧化反应(HzOR,N2H4+4OH-→N2+4H2O+4e-,-0.33 V(vs RHE))因其较低的热力学氧化电势而受到关注[15]。它以超低电压辅助OWS不仅能够有效降低制氢耗能,而且催化产物是N2和H2,可避免H2/O2混合产生的安全问题,在移动式储氢等装置中具有潜在的应用前景[16-20]。因此,HzOR相关研究引起了人们的关注。例如,Shao课题组通过碱辅助合成和H2还原途径制备了具有Rh-O-Rh界面的Rh/RhOx纳米片,HzOR测试中所需要的过电势η10为-28 mV,组成的电解池所需电压E10为63 mV[7]。还有研究者通过电沉积法将钌单原子固定在海胆状三氧化钨的氧空位中,得到的CC@WO3/Ru SAs催化剂在HzOR和HzOR辅助的OWS测试中,仅需-58 mV的过电势和25 mV的电压就可达到10 mA·cm-2的电流密度[21]。尽管取得了一些进展,但仍存在一些关键问题需要解决。例如,当前已报道的电催化HzOR的工作电位仍远远高于理论值,从而导致HzOR辅助的OWS的电解电压较高。此外,由于HER和HzOR活性中间体的不同,兼具电催化HzOR和HER能力的双功能电催化剂开发还很少。因此,需要综合平衡HER水解离的决速步骤和HzOR脱氢的动力学步骤,以实现优异的电催化HzOR和HER双功能性[19-25]。
近年来,钌(Ru)基催化剂由于其对HER/OER的高催化活性和较好的稳定性已引起人们广泛的关注。它不仅具有较好的成本优势(约为Pt的4%),还具有与铂相似的氢键能(Ru—H,272.08 kJ·mol-1),被认为是铂的竞争替代品[26-29]。虽然单一的Ru对水的吸附和裂解性能相对较弱,但其有利于氢中间体的吸附和氢气的脱附[30]。特别是,Ru位点还可以稳定HzOR中间体,降低决速步的能垒,有利于促进HzOR过程[19]。因此,进一步开发双功能钌基催化剂十分必要。
另一方面,低成本的钼基催化剂(MoO3、MoO2、MoS2)被 用 作HER催 化 剂[2,31-33]。然 而,纯MoO3、MoO2、MoS2的本征电催化活性较低。为了提高其活性,人们通过空位策略或与金属杂化来进一步优化性能。例如,通过软模板法合成了富含氧空位的介孔MoO3-x纳米结构,丰富的氧空位和中空结构使其具有优异的HER性能[31]。至今为止,将富含氧空位的氧化钼和Ru金属杂化应用于HzOR及其辅助OWS的催化剂还未见到报道。我们采用静电相互作用和程序热还原策略,制备了Ru团簇均匀锚定于富含氧空位的MoO3-x纳米带的双功能催化剂(Ru/MoO3-x)。当应用于HzOR时,Ru/MoO3-x催化剂的过电势η10为-79 mV,Tafel斜率为16 mV·dec-1,循环10 000圈后稳定性仍较好,明显优于单一MoO3-x和商业化20%Pt/C。对于HER,该催化剂表现出与商业化20% Pt/C相近的性能,过电势η10和Tafel斜率分别为-27 mV和39 mV·dec-1。由该催化剂构成的电解池仅需13 mV就可达到10 mA·cm-2的电流密度,显著优于Pt/C和已报道的大多数催化剂。本工作为HzOR辅助OWS双功能催化剂的设计和合成提供了一种新策略。
1 实验部分
1.1 试剂
试剂包括(NH4)6Mo7O24·4H2O(分析纯99%,国药集团化学试剂有限公司)、RuCl3(优级纯40%,国药集团化学试剂有限公司)、硝酸(HNO3,国药集团化学试剂有限公司)、20%Pt/C(Alfa Aesar化学有限公司)、CrCl3(分析纯98%,Alfa Aesar化学有限公司)、5%萘酚(Alfa Aesar化学有限公司)、无水乙醇(国药集团化学试剂有限公司)。实验过程中使用的溶剂水均为去离子水,所有化学品和试剂在使用前均没有进一步纯化。
1.2 催化剂的制备
1.2.1 MoO3的合成
根据文献方法改变反应温度合成了MoO3[34]。具体如下:称取0.5 g(NH4)6Mo7O24·4H2O溶于25 mL去离子水中,依次加入5 mL浓HNO3和50 mg CrCl3(形貌导向剂)。将混合溶液置于磁力搅拌器上搅拌30 min后转移到100 mL反应釜中,在200℃下反应3 h,冷却至室温后用去离子水洗涤2次,产物冷冻干燥。
1.2.2 Ru/MoO3-x的合成
通过两步法制得Ru/MoO3-x-600,具体如下:称取20 mg MoO3溶于10 mL去离子水中,超声10 min使其分散均匀,得到白色胶体溶液,随后加入600 μL RuCl3溶液(深棕色,4.5 mg·mL-1),搅拌2 h后离心分离(上清液呈无色),产物冷冻干燥(棕色)。接下来将上述产物置于管式炉中,在5% H2/Ar气氛下450℃还原2 h,即可得到产物(即Ru/MoO3-x-600,600代表RuCl3投料量)。
保持其余实验条件不变,改变RuCl3投料量至200和1 000 μL,即可分别得到Ru/MoO3-x-200和Ru/MoO3-x-1000。
1.3 仪器表征
采用透射电子显微镜(TEM,加速电压200 kV,JEM-200CX,日本)、高分辨透射电子显微镜(HRTEM,加速电压200 kV,JEOL-2100F)和扫描电子显微镜(SEM,加速电压200 kV,JEOL-2100F)观察样品形貌;采用电感耦合等离子体(ICP,jarrell-Ash 1100+2000)、阳极转靶X射线衍射仪(XRD,D/max 2500VL/PC,CuKα激发源,λ=0.154 060 nm,扫描范围10°~90°,工作电压45 kV,电流200 mA,日本)测试元素组成和结构;采用X射线光电子能谱(XPS,测试管电压和电流分别是15 kV和10 mA,PHI 5000 Verasa,Al激发源,ULAC-PHI,Inc.)测试元素价态,测试得到的所有数据以C1s峰(284.8 eV)为标准校正。
1.4 电化学测试
所有的电化学测试均在上海辰华仪器公司的CHI 660E电化学工作站上进行,标准的三电极测试体系用于HER和HzOR测试,负载催化剂的碳布、高纯石墨棒和Hg/HgO电极(1 mol·L-1KOH)分别为工作电极、对电极和参比电极。工作电极制备方法如下:取一定量催化剂粉末溶于乙醇、水、萘酚的混合溶液(体积比为4.5∶4.5∶1),超声30 min~1 h得到均匀溶液。取250 μL催化剂溶液均匀涂抹于碳布电极(1 cm×1 cm)表面,真空干燥后即可得到负载催化剂的碳布工作电极,催化剂载量500 μg·cm-2。电化学测试之前,首先通过循环伏安(CV)法进行预处理,消除任何可能的表面污染物使电位稳定。然后在-1.2~-0.8 V的电压下进行CV测试,在-1.4~-0.8 V的电压窗口获得极化曲线(LSV,有IR校正),扫速5.0 mV·s-1。所有电位均根据公式E(vs RHE)=E(vs Hg/HgO)+0.095+0.059pH转换为相对RHE电位。HzOR辅助的OWS实验在H型双电极电解池中进行,两侧分别填充1.0 mol·L-1KOH+0.5 mol·L-1N2H4和1.0 mol·L-1KOH溶液。负载催化剂的碳布作为阴极和阳极。电化学双电层电容(Cdl)的CV测试在非法拉第区域进行,电压范围为0.124~0.224 V(vs RHE)。不同催化剂的电化学活性表面积(ECSA)通过ECSA=Cdl/Cs计算得到(Cs为催化剂的比电容)。电化学阻抗(EIS)测试电位η=100 mV,频率100 kHz~0.1 Hz。
2 结果与讨论
2.1 催化剂合成与表征
首先对制得的MoO3进行ζ电位测试(图1a),结果显示MoO3溶液的电位为-43 mV,表明其具有负电性。因此,将RuCl3和MoO3溶液混合,由于静电相互作用,Ru3+可充分吸附在MoO3上,吸附前后溶液的颜色变化如图1b所示。对吸附后的样品进行ζ电位测试,其电位下降至-27 mV,进一步证实了Ru3+离子较好地吸附在MoO3表面。将获得的Ru3+/MoO3在H2/Ar中热还原,即可制得Ru/MoO3-x-600。图1c是MoO3和Ru/MoO3-x-600的XRD图。图中12.8°、23.4°、25.7°、27.4°、29.1°和33.8°处的衍射峰对应单斜MoO3(PDF No.05-508)的(020)、(110)、(040)、(021)、(130)和(111)晶面。Ru/MoO3-x-600位于37.5°、42.2°和43.4°处的衍射峰可归属于六方Ru(PDF No.88-1734)的(100)、(002)和(101)晶面,但该图中未显现MoO3-x氧化物的衍射峰,这表明Ru/MoO3-x-600中MoO3-x可能是以无定形形式存在。通过SEM、TEM和HRTEM对Ru/MoO3-x-600的形貌和微结构进行了分析(图1d、1e)。从SEM图(图1d)中可以观察到Ru/MoO3-x-600是长度达数微米的纳米带。TEM图(图1e)显示纳米带的宽度约为200 nm。特别地,从该图中未见有较大尺寸的颗粒,表明超小Ru纳米团簇均匀锚定在MoO3-x上。进一步的证据来自于HRTEM的表征(图1f和图S1,Supporting information),图中晶面间距0.23和0.33 nm分别归属于Ru的(100)晶面和MoO3-x的(012)晶面;Ru纳米团簇为(1.4±0.2)nm(图S2),均匀锚定在MoO3-x上(图S3)。值得注意的是,在HRTEM图中可以清楚地观察到MoO3-x晶格长程无序,表明以无定形形式存在,这与XRD分析的结果一致。此外,从图中还可以看到Ru/MoO3-x-600中存在大量缺陷和位错,这可以作为电催化的活性中心,有利于电催化性能的提升。ICP测试表明,该样品中Ru的质量分数为4.25%,含量较少。综上分析可知,通过Ru3+和MoO3的静电相互作用和随后的程序热还原,可以方便地制得超小Ru纳米团簇均匀锚定分布在MoO3-x的异质纳米结构。
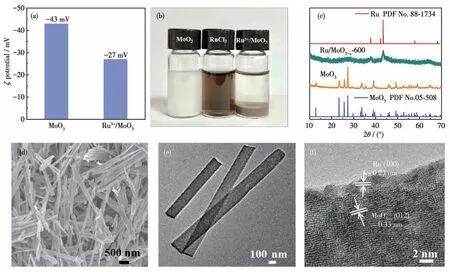
图1 (a)MoO3和Ru3+/MoO3的ζ电位对比;(b)MoO3、RuCl3和Ru3+/MoO3粉末分散在水中的照片;(c)MoO3和Ru/MoO3-x-600的XRD图;Ru/MoO3-x-600的(d)SEM图、(e)TEM图和(f)HRTEM图Fig.1(a)Comparison of ζ potential of MoO3 and Ru3+/MoO3;(b)Pictures of MoO3,RuCl3,and Ru3+/MoO3 powder dispersed in water;(c)XRD pattern of MoO3 and Ru/MoO3-x-600;(d)SEM image,(e)TEM image,and(f)HRTEM image of Ru/MoO3-x-600
为了解Ru/MoO3-x-600的存在形态及组分间相互作用,对其进行了XPS分析。如图2a所示,XPS全谱图显示了Ru/MoO3-x-600中Ru、Mo和O元素的存在。从Ru/MoO3-x-600的O1s精细XPS谱图(图2b)中可知,除出现Mo—O键(530.9 eV)和O—H键(532.5 eV)外,还出现氧空位的信号(531.5 eV),含量高达47%,而MoO3的氧空位含量仅为16%,表明H2的还原导致了催化剂中氧空位含量显著增多。图2c为Ru/MoO3-x-600和MoO3的Mo3d精细XPS谱图,由图可知,MoO3中Mo主要以Mo6+形式存在,而Ru/MoO3-x-600中同时存在Mo6+和Mo5+。这是因为H2在还原Ru3+的同时,也将部分Mo6+还原为Mo5+。此外,
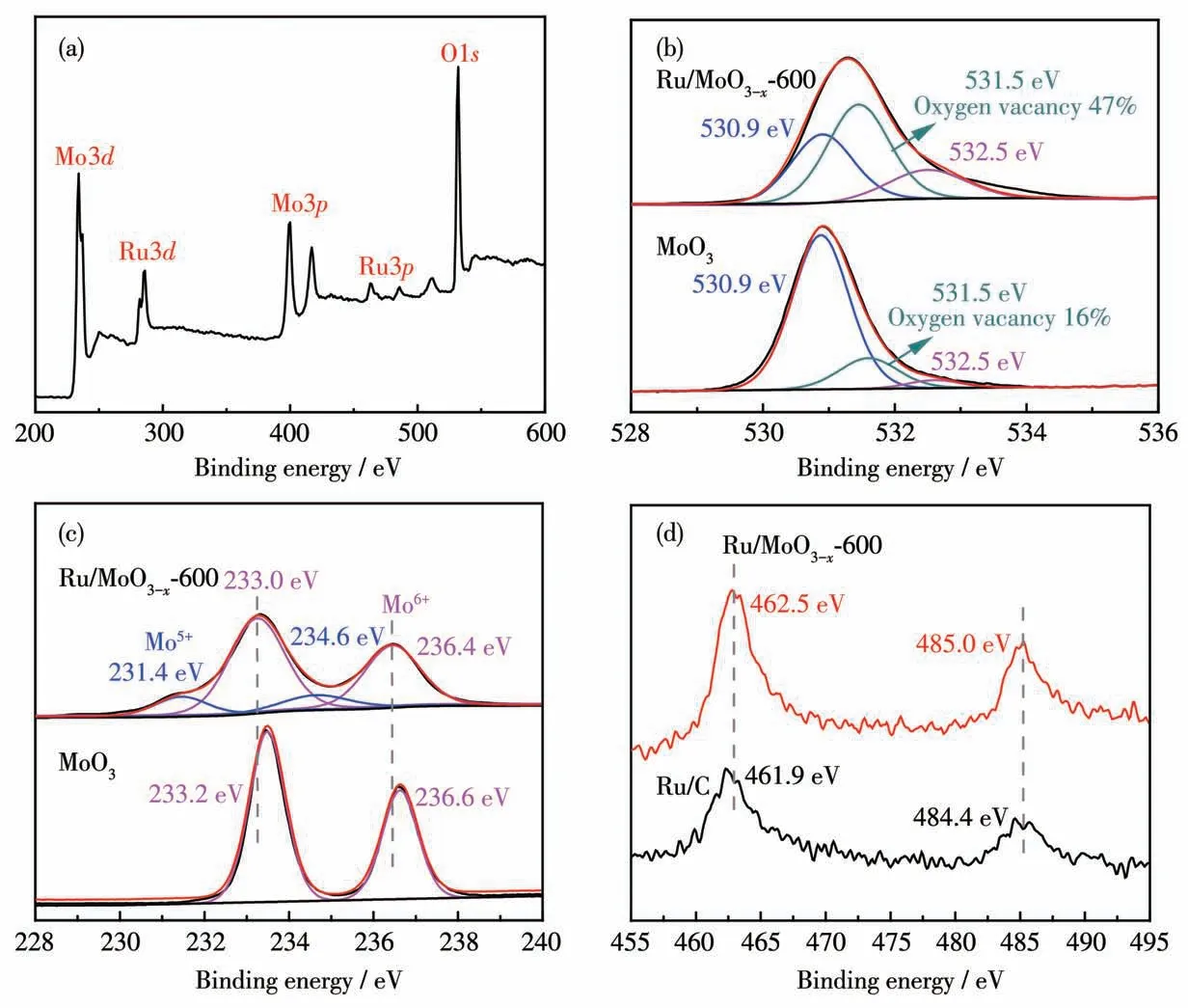
图2 (a)Ru/MoO3-x-600的XPS全谱图;Ru/MoO3-x-600和MoO3的(b)O1s谱图和(c)Mo3d谱图;(d)Ru/MoO3-x-600和Ru/C的Ru3p谱图Fig.2(a)XPS survey spectrum of Ru/MoO3-x-600;(b)O1s and(c)Mo3d spectra of Ru/MoO3-x-600 and MoO3;(d)Ru3p spectra of Ru/MoO3-x-600 and Ru/C
相比于MoO3,Ru/MoO3-x-600的Mo3d峰向低结合能方向移动了0.2 eV,说明Ru和MoO3-x之间存在电子相互作用。图2d是Ru/MoO3-x-600和Ru/C的Ru3p精细XPS谱图,462.5和485.0 eV处的峰分别归属于Ru3p3/2和Ru3p1/2。相 比 于Ru/C,Ru/MoO3-x-600的Ru3p峰向高结合能方向偏移,这可以归因于电子从Ru向O的转移[35],这将改变Ru/MoO3-x异质界面的电荷密度,从而优化其电子结构[36]。
2.2 催化剂的性能
2.2.1 Ru/MoO3-x-600的HzOR性能
以电催化HzOR为探针反应,在典型的三电极体系中测试了Ru/MoO3-x-600的催化性能。作为比较,还测试了单组分MoO3-x、商业化20% Pt/C和5%Ru/C以及不同Ru源用量的Ru/MoO3-x-200和Ru/MoO3-x-1000的催化性能。图3a为上述不同催化剂的HzOR LSV曲线。由图中可知,Ru/MoO3-x-600仅需-79 mV的过电势就可达到10 mA·cm-2的电流密度,明显优于Ru/MoO3-x-200(-57 mV)、MoO3-x(339 mV)、Pt/C(-1 mV)、Ru/C(-62 mV)以及大多数目前报道的催化剂(图3e和表S1)。图3b给出了不同催化剂的Tafel斜率。Ru/MoO3-x-600的Tafel斜率为16 mV·dec-1,显 著 小 于Ru/MoO3-x-200(92 mV·dec-1)、MoO3-x(153 mV·dec-1)、Ru/C(34 mV·dec-1)和Pt/C(44 mV·dec-1),表明Ru/MoO3-x-600的HzOR催化动力学性能更优异。Feng课题组报道了Ru团簇负载于N掺杂介孔含氧碳球(Ru10@mONC)用于催化HzOR,其Tafel斜率为14.2 mV·dec-1。密度泛函理论(DFT)计算表明*N2H2→*N2H为HzOR脱氢过程的决速步[24]。本工作所制得的Ru/MoO3-x-600的Tafel斜率为16 mV·dec-1,与Ru10@mONC的Tafel斜率相近。据此,我们推测Ru/MoO3-x-600催化HzOR脱氢的决速步可能是*N2H2→*N2H。确切的催化机理需要进一步深入的研究。
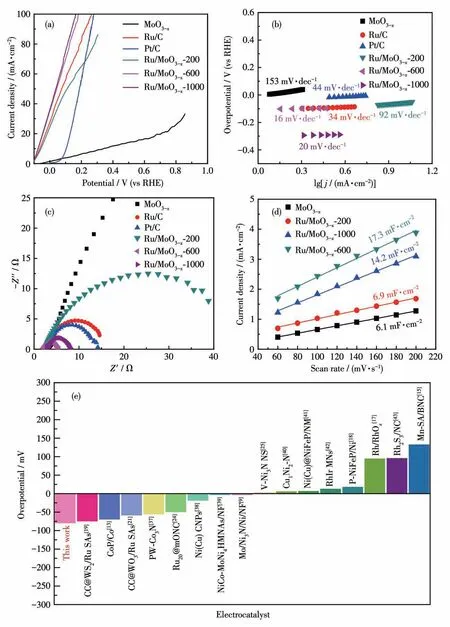
图3 MoO3-x、Ru/C、Pt/C、Ru/MoO3-x-200、Ru/MoO3-x-600和Ru/MoO3-x-1000在0.5 mol·L-1 N2H4+1.0 mol·L-1 KOH溶液里的(a)LSV曲线、(b)Tafel斜率、(c)EIS谱图和(d)Cdl;(e)与最近报道的其他电催化剂HzOR性能的比较Fig.3(a)LSV curves,(b)Tafel slopes,(c)EIS spectra,and(d)Cdl of MoO3-x of Ru/C,Pt/C,Ru/MoO3-x-200,Ru/MoO3-x-600,and Ru/MoO3-x-1000 in 0.5 mol·L-1 N2H4+1.0 mol·L-1 KOH;(e)Comparison of HzOR performance with other recently reported electrocatalysts
我们还进一步测试了上述催化剂的电化学阻抗和电化学双层电容(图3c、3d)。由图3c可知,Ru/MoO3-x-600的界面电子转移电阻(5.5 Ω)明显小于Pt/C(15.0 Ω)、Ru/C(17.0 Ω)和MoO3-x(150.0 Ω),说明Ru/MoO3-x-600具有更快的电子转移速率。此外,由图3d可知,Ru/MoO3-x-600的电化学双层电容值(17.3 mF·cm-2)也 优 于Ru/MoO3-x-200(6.9 mF·cm-2)、Ru/MoO3-x-1000(14.2 mF·cm-2)和MoO3-x(6.1 mF·cm-2),表明Ru/MoO3-x-600具有更多电化学活性位点。
上述测试表明,和其它催化剂相比,Ru/MoO3-x-600催化剂具有最优异的HzOR催化活性。究其原因,主要可归结于下列因素:Ru位点可以有效吸附N2H4,降低肼脱氢的能垒,是HzOR的反应活性中心[19];而且Ru团簇的超小尺寸及其在MoO3-x纳米带表面的均匀分布和Ru/MoO3-x的异质结构有利于暴露更多的活性位点;此外,MoO3-x丰富的氧空位优化了界面电荷分布,从而改善了电子转移动力学[31]。对于Ru/MoO3-x-1000具有类似于Ru/MoO3-x-600的电催化HzOR活性的原因,可能是Ru源用量的增加导致电催化活性位点增多,性能得到提升;但是当Ru源投料量增加到1 000 μL时,过多的用量使得Ru团簇聚集和变大,妨碍活性位点的暴露,从而影响了催化活性的进一步提升。
稳定性是影响催化剂实际应用的重要参数之一。因此,对Ru/MoO3-x-600进行了HzOR稳定性测试。如图S4所示,连续4 000圈CV循环后,其电流密度几乎没有变化;10 000圈CV循环后,电流只有轻微衰减。计时电流测试结果表明在24 h持续测试下,其电流也相对比较稳定(图S5),表明Ru/MoO3-x-600具有较好的催化稳定性。对稳定性测试后的Ru/MoO3-x-600进行了TEM、HRTEM和XRD表征。从TEM图(图S6a)可以看出,Ru/MoO3-x-600的纳米带形貌几乎没有变化。HRTEM图像(图S6b)和XRD图(图S7)表明稳定性测试后,Ru/MoO3-x-600的组成和物相没有发生明显变化。综合以上分析可知,Ru/MoO3-x-600催化剂具有优异的HzOR催化活性和较好的稳定性。
2.2.2 Ru/MoO3-x-600的HER性能
为了构建肼辅助的全解水电解池,在碱性介质中对Ru/MoO3-x-600的HER性能进行了测试。从图4a给 出 的HER LSV曲 线 可 知,Ru/MoO3-x-600的HER性能优于Ru/MoO3-x-200和Ru/MoO3-x-1000,仅需要-27 mV的工作电位就可达到10 mA·cm-2电流密度,显著优于MoO3-x,可与Pt/C和Ru/C相媲美。进一步通过Tafel斜率评价了HER的电催化动力学。从图4b可以看出,Ru/MoO3-x-600的Tafel斜率是39 mV·dec-1(与Pt/C的36 mV·dec-1相接近),表明Volmer-Heyrovsky机理主导着HER过程。对比MoO3-x的Tafel斜率(162 mV·dec-1),Ru/MoO3-x-600的Tafel斜率显著降低,这可能是因为Ru与MoO3-x的异质化调节了催化剂表面的电子密度,降低了水吸附和脱氢的能垒[29]。此外,Ru/MoO3-x-600还具有良好的电催化稳定性,10 000圈CV循环后,HER LSV曲线几乎保持不变(图S8)。24 h连续的计时电流测试进一步证实了Ru/MoO3-x-600长久的电催化稳定性(图S5)。
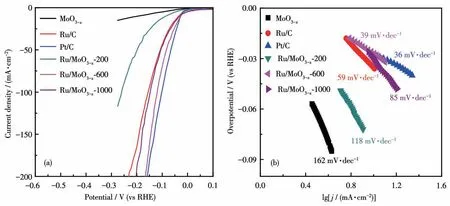
图4 MoO3-x、Ru/C、Pt/C、Ru/MoO3-x-200、Ru/MoO3-x-600和Ru/MoO3-x-1000在1.0 mol·L-1 KOH溶液里的(a)LSV曲线和(b)Tafel斜率Fig.4(a)LSV curves and(b)Tafel slopes of MoO3-x,Ru/C,Pt/C,Ru/MoO3-x-200,Ru/MoO3-x-600,and Ru/MoO3-x-1000 in 1.0 mol·L-1 KOH
2.2.3 HzOR辅 助OWS的Ru/MoO3-x-600双 功 能性能
鉴于Ru/MoO3-x-600优异的电催化HzOR和HER双功能性,进一步将其组装用于肼辅助的OWS。结果如图5a所示,Ru/MoO3-x-600//Ru/MoO3-x-600电解池仅需13 mV的电压就可达到10 mA·cm-2电流密度。相应地,Pt/C//Pt/C电解池需要154 mV的工作电压,说明作为双功能催化剂,Ru/MoO3-x-600的性能优于Pt/C。更进一步,上述电压还优于目前报道的大多数催化剂(图5d和表S2)。作为对比,我们还在不含肼的1.0 mol·L-1KOH电解液中进行了OWS测试。结果表明,Ru/MoO3-x-600//Ru/MoO3-x-600电解池需要1.486 V的高电压才能达到10 mA·cm-2电流密度,充分表明HzOR辅助的OWS可明显减少电能的消耗,提高OWS的产氢效率。图5b是H型双电极电解池HzOR辅助OWS的装置图,由图可以清楚地观察到阴极和阳极均产生了大量气泡,分别是H2和N2。此外,Faradic实验结果表明,HzOR辅助OWS的产氢速率为0.2 mmol·h-1,产氢值与理论值较好地吻合,制氢效率接近100%(图S9)。采用计时电位法测定了该电池的稳定性,在经过24 h持续测试后,Ru/MoO3-x-600//Ru/MoO3-x-600的电压仍比较稳定,说明该催化剂具有优异的稳定性(图5c)。
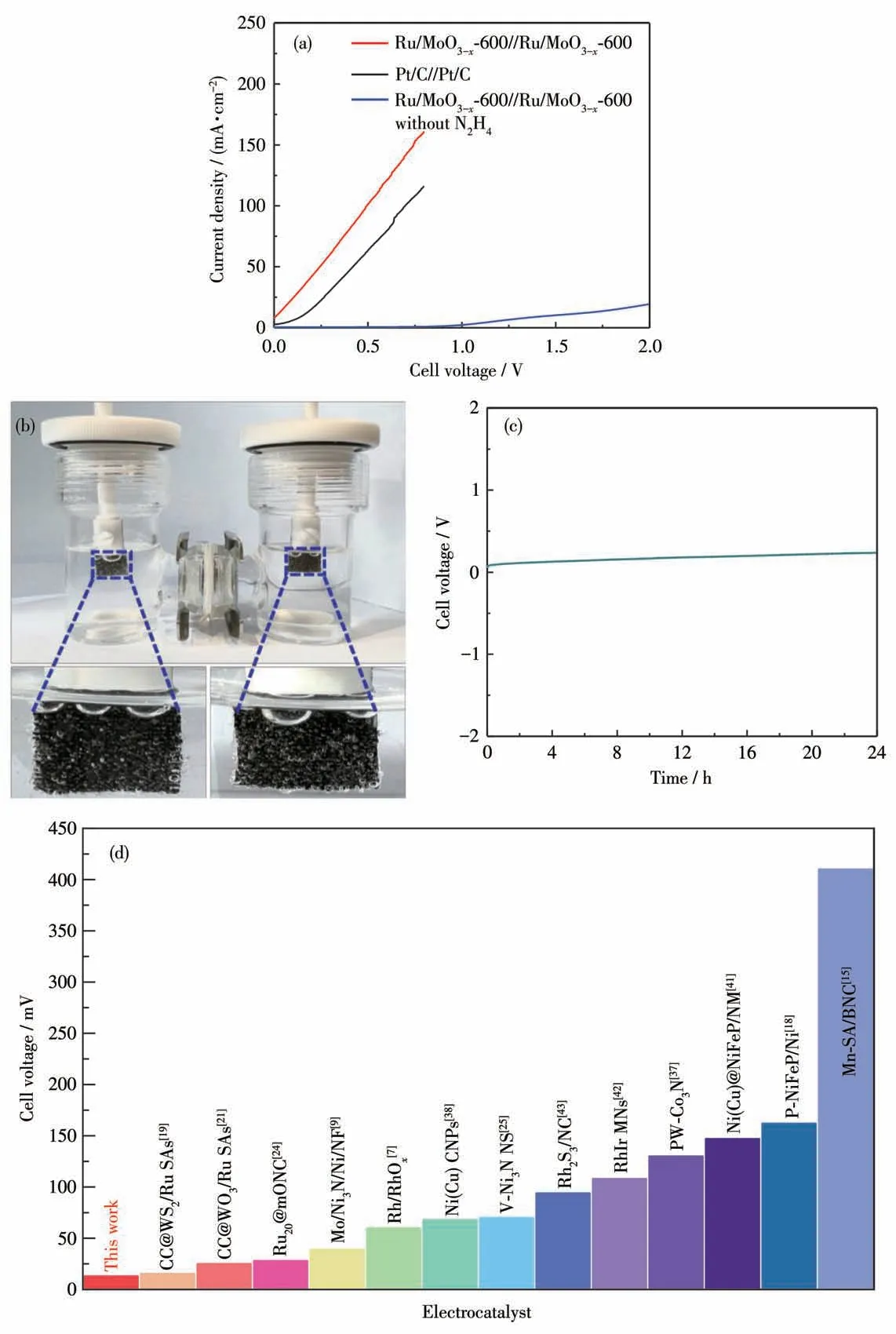
图5 (a)Ru/MoO3-x-600//Ru/MoO3-x-600双电极在有无肼条件下OWS的LSV曲线;(b)肼辅助全解水电解池构造的光学图像;(c)在0.5 mol·L-1 N2H4+1.0 mol·L-1 KOH中,Ru/MoO3-x-600//Ru/MoO3-x-600的HzOR辅助OWS的长期稳定性测试;(d)与最近报道的其他电催化剂HzOR辅助OWS性能的比较Fig.5(a)LSV curves of two-electrode OWS for Ru/MoO3-x-600//Ru/MoO3-x-600 with or without hydrazine;(b)Optical image of HzOR assisted-OWS cell;(c)Long-time stability test of HzOR assisted-OWS for Ru/MoO3-x-600//Ru/MoO3-x-600 cell in 0.5 mol·L-1 N2H4+1.0 mol·L-1 KOH;(d)Comparison of HzOR assisted OWS performance with other recently reported electrocatalysts
3 结论
巧妙地利用MoO3纳米带表面带负电的特性,通过简单的静电相互作用和随后的程序热还原法,方便地制得了Ru团簇均匀锚定于富含氧空位的MoO3-x纳米带双功能催化剂。该催化剂在碱性条件下表现出优异的电催化HER和HzOR双功能性;更进一步,由该催化剂构建的HzOR辅助OWS电解池仅需13 mV的电压即可驱动10 mA·cm-2电流密度,显著优于商业化20% Pt/C及目前报道的大部分催化剂。其优异的性能可归纳为以下因素的综合影响:(1)Ru团簇的超小尺寸及其在MoO3-x纳米带表面的均匀分布可较大程度暴露活性位点;与此同时,Ru/MoO3-x异质结构创造了大量界面缺陷,提升了活性位点数量;(2)Ru/MoO3-x的异质结构和MoO3-x丰富的氧空位优化了界面电荷分布,改善了电子转移动力学;(3)Ru和MoO3-x之间的协同作用,不仅有利于降低水吸附、裂解和氢析出的能垒,还可以促进N2H4的吸附和脱氢过程。这种纳米团簇、氧空位和异质结构共优化的设计策略不局限于Ru和MoO3-x,还可以被推广到其它具有特定性质的优选组分,对新型、高效多功能电催化剂的开发和应用具有重要意义。
Supporting information is available at http://www.wjhxxb.cn
