吲哚酚类糖苷酶显色底物6-氯-3-吲哚-β-D-葡萄糖苷的合成工艺研究
2022-10-10李新章周清浩范高福胥振国
尹 伟,胡 琪,李新章,周清浩,范高福,胥振国
(1.合肥职业技术学院 生物工程学院,安徽 合肥 238000;2.南通睿沣新材料技术有限公司 化学成分分析中心,江苏 南通 226000;3.中国科学技术大学高分子科学与工程系 中科院软物质化学重点实验室,安徽 合肥 230026)
近年来, 由病原微生物导致的疾病, 如禽流感(H7N9、H5N1)、非典型肺炎(SARS)以及目前仍在全球肆虐的新型冠状病毒(2019-nCoV)等所感染的疾病,传染性都极强,通常能够造成世界性的大流行[1-4],因此在对病原微生物的检测方面,务必做到精准、快速、高效和简便。 传统的病原微生物检测方式具有一定的局限性,检测过程较为繁琐,检测持续周期长,亦可能出现一定的误差。
显色培养基是指在传统的微生物分离培养基中加入了一类特殊的化学合成底物的新型培养基,在微生物培养过程中, 通过微生物代谢产生特异性的酶而显色,最终根据不同颜色对各菌群进行鉴别[5-7]。培养基显色底物主要有4-甲基伞形酮类、吲哚酚类等,此方法在微生物的检测应用上,有效提高了病原微生物检测的准确性,同时又大大缩短了检测时间,应用前景十分广泛。
有研究报道[8-9],利用苯胺、卤代物、N-(2-羧基)苯基甘氨酸及2-氯-6-氟苯甲醛等作为起始合成原料合成吲哚酚类显色底物,终产物的收率、纯度以及反应时间均存在一定的不足。 本研究在前期实验基础上, 以常见试剂邻氨基苯甲酸为原料合成吲哚酚类糖苷酶显色底物6-氯-3-吲哚-β-D-葡萄糖苷,通过卤化等反应得到目标产物, 整体收率有大幅度的提升,显现出很多优点:(1)初始原料邻氨基苯甲酸价格低廉,容易购买;(2)合成路线步骤相对较少;(3) 实验过程中涉及的易燃易爆有毒等化学试剂较少,相对环保。 上述多方面的合成优点为此显色底物的综合利用提供了强有力的保障。 同时,实验中对所有合成中间体进行了结构表征,优化了该检测底物的合成工艺路线,以期高效简便地合成此目标化合物。
1 合成路线的选择
吲哚酚类显色底物的合成改造主要集中在苯环上, 一般都是在这些位置引入卤素或硝基等进行修饰,以达到既保证其具有较好的显色效果,又降低合成的原料成本,提高目标物收率的目的。 在参考了相关文献[10]之后,最终确定了合成方案,见图1。
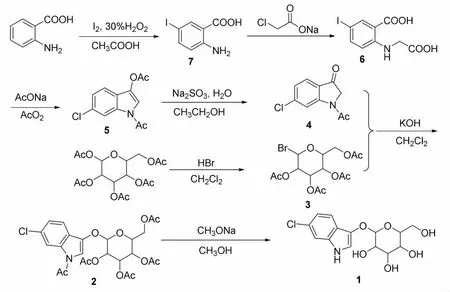
图1 显色底物6-氯-3-吲哚-β-D-葡萄糖苷的合成工艺路线
2 实验部分
2.1 试剂与仪器
(1)实验试剂:邻氨基苯甲酸[西亚化学科技(山东)有限公司],冰醋酸(河北鸿韬生物工程有限公司),双氧水(安德化工有限公司),NaHSO3(宜兴市锦程化工有限公司),NaOH(四川佰春科技有限责任公司),氯乙酸钠(淄博鲁硕化工有限公司),碳酸钠(济南元素化工有限公司),盐酸(国药集团化学试剂有限公司),乙酸钠(国药集团化学试剂有限公司),乙酸酐(国药集团化学试剂有限公司),乙醇(济宁博诚化工有限公司),CH2Cl2(济南启辰化工有限公司),无水硫酸镁(国药集团化学试剂有限公司),无水硫酸钠(国药集团化学试剂有限公司),CH3ONa(国药集团化学试剂有限公司),CH3OH(济宁博诚化工有限公司),乙酸乙酯(山东辰宇化工有限公司)。
(2)实验仪器:旋转蒸发仪(RE-2000A,上海亚荣生化仪器厂),电子天平(MF1035C,济南爱来宝仪器设备有限公司),真空干燥箱(DZF-6210A,苏州纳美瑞电子科技有限公司),集热式加热搅拌器(DF-101S,上海梅颍浦仪器仪表制造有限公司),紫外灯(ZF-2,杭州齐威仪器有限公司),循环水真空泵(SHZIII,上海亚荣生化仪器厂),硅胶柱(100~200 目,200~300 目,青岛海洋化工有限公司)。
2.2 实验过程
2.2.1 化合物7(5-碘-2-氨基苯甲酸)的合成
在100 mL 的三颈烧瓶中, 加入邻氨基苯甲酸2.5 g(0.018 mol)、冰醋酸30 mL,搅拌溶解,随后加入2.3 g(0.009 mol)碘粒,继续搅拌,反应液呈深棕色, 待烧瓶中固体完全溶解时, 滴加双氧水0.31 g(0.009 mol,30%),在20 min 内滴加完毕,反应体系升温至60 ℃,反应3.0 h。 每隔0.5 h,用TLC 点板跟踪反应情况。待反应完成后,将烧瓶中反应产物缓慢倒入NaHSO3(1.0 mol/L,50 mL)中,继续搅拌1.5 h,析出黄色固体,抽滤、用水洗涤3 次,最终得到黄色固体,收率达95.8%。1H-NMR(500 MHz,DMSO-d6)δ:7.90(d,J=2.2 Hz,1H),7.51(dd,J=8.5,2.2 Hz,1H),6.64(d,J=8.3 Hz,1H)。
2.2.2 化合物6(5-碘-2-羧基甲基氨基苯甲酸)的合成
在100 mL 的三颈烧瓶中加入5-碘-2-氨基苯甲酸2.5 g(0.01 mol)、NaOH 0.4 g(0.01 mol)及H2O 10.0 mL,搅拌0.5 h 至固体溶解,溶液澄清。 同时在烧杯中加氯乙酸钠1.33 g(0.012 mol)、碳酸钠1.0 g(0.012 mol)及H2O 30 mL,不断搅拌至全溶解,随后用恒压滴液漏斗缓慢滴加至三颈烧瓶中,0.5 h滴加完毕,升温至80 ℃,恒温反应18 h,在反应过程中,用TLC 点板跟踪反应情况,直至原料点没有变化为止,停止反应。使用10%盐酸调pH 值(pH=3.0),过滤重结晶得到白色固体, 收率达60.6%。1H-NMR(500 MHz,DMSO-d6) δ:8.00 (d,J=2.1 Hz,1H),7.48(dd,J=8.7,2.1 Hz,1H),6.46(d,J=9.1 Hz,1H),3.96(s,2H)。
2.2.3 化合物5(N-乙酰基-5-碘吲哚-3-乙酸酯)的合成
在100 mL 的三颈烧瓶中加入5-碘-2-羧基甲基氨基苯甲酸2.5 g(0.008 mol)、乙酸酐20.0 mL,搅拌溶解,在持续搅拌状态下加入乙酸钠2.9 g(0.07 mol),随后反应升温至125 ℃,搅拌、回流反应4.0 h。 反应无气泡产生时,停止反应。 在搅拌下加入冰水,有固体产生,继续搅拌1.0 h 后,在4 ℃下放置过夜,抽滤得到深棕色固体。用硅胶柱纯化[V(乙酸乙酯)∶V(石油醚)=1∶6], 得固体收率为67.8%。1H-NMR(500 MHz,DMSO-d6) δ:8.13(d,J=8.5 Hz,1H),7.90(d,J=1.4 Hz,1H),7.11 (dd,J=8.5,1.8 Hz,1H),2.63 (s,3H),2.35(s,3H)。
2.2.4 化合物4(N-乙酰基-6-氯-3 吲哚酮)的合成
在100 mL 的三颈烧瓶中加N-乙酰基-6-氯吲哚-3-乙酸酯1.5 g(0.006 mol)、乙醇20 mL,搅拌至固体完全溶解,同时将Na2SO31.83 g(0.015 mol)溶于15.0 mL H2O 中,缓慢滴加至三颈烧瓶中,0.5 h 滴加完毕,随后升温至回流,反应8 h。 反应过程中用TLC 点板跟踪反应情况。 反应结束时,将溶液转移至单口瓶中,减压浓缩除去乙醇,加入乙酸乙酯萃取3次,用无水硫酸钠干燥,最终得到固体产物,收率达87.2%。1H-NMR(500 MHz,DMSO-d6)δ:8.42(s,1H),7.65 (d,J=7.9 Hz,1H),7.30 (dd,J=8.1,1.7 Hz,1H),4.62(s,2H),2.25(s,3H)。
2.2.5 化合物3(溴代四乙酰葡萄糖)的合成
向250 mL 的三颈烧瓶中加入五-O-乙酰基-β-D-吡喃葡萄糖5.0 g(0.013 mol)、干燥CH2Cl2100 mL,搅拌溶解,整个过程于冰浴中进行,将HBr 的冰醋酸溶液(45%,30 mL)缓慢滴加至反应瓶中,滴加完毕后, 室温搅拌12 h, 整个反应过程用TLC 点板跟踪反应情况。反应结束后,加入CH2Cl2(50 mL),用饱和NaHCO3溶液洗涤5 次。 分出有机层,用无水硫酸镁干燥,过滤,减压浓缩,得淡棕色油状物,用石油醚及乙醚混合溶剂重结晶, 得到白色固体, 收率达90.3%。1H-NMR(500 MHz,CDCl3) δ:6.60(d,J=3.91 Hz,1H),5.51(t,1H),5.22 (t,1H),4.83 (t,1H),4.31(m,2H),4.15(m,1H)。
2.2.6 化合物2 [(1-乙酰基-6-氯吲哚-3-基)-2,3,4,6-四乙酰基-β-D-葡萄糖苷]的合成
在N2保护及冰浴条件下, 向100 mL 的三颈烧瓶中加入N-乙酰基-6-氯-3-吲哚酮1.00 g(0.005 mol)、NaNH20.2 g(0.005 mol)及CH2Cl250 mL,搅拌1 h。用CH2Cl2(30 mL)溶解溴代四乙酰葡萄糖3.93 g(0.01 mol),滴加至三颈瓶中,室温搅拌反应12 h,整个反应过程用TLC 点板跟踪反应情况。 反应结束后分液,用无水硫酸钠干燥,使用柱色谱法[V(石油醚)∶V(乙酸乙酯) =3∶1]纯化及乙醇重结晶,得到白色固体,收率达48.9%。1H-NMR (500 MHz,DMSOd6) δ:8.32(s,1H),7.51(s,1H),7.42(d,J=8.2 Hz,1H),7.35(dd,J=8.0,2.1 Hz,1H),5.51(d,J=8.2 Hz,1H),5.42(t,J=9.7 Hz, 1H),5.15(dd,J=9.9,8.2 Hz,1H),5.08 (t,J =9.7 Hz,1H),4.09 ~4.25 (m,3H),2.65(s,3H),2.02(dd,J=19.0,17.4 Hz,12H)。
2.2.7 化合物1 (6-氯-3-吲哚-β-D-葡萄糖苷)的合成
向100 mL 的三颈烧瓶中加入(1-乙酰基-6-氯吲哚-3-基)-2,3,4,6-四乙酰基-β-D-葡萄糖1.0 g(0.002 mol)、无水CH3OH 40 mL,搅拌1 h,随后加入KOH 0.1 g(0.002 mol),室温搅拌反应4.0 h,溶液浓缩,有固体产生,抽滤,用甲醇洗涤,干燥,使用硅胶柱色谱法(纯乙酸乙酯)纯化,最终得到产物,收率达90.3%。1H-NMR (500 MHz,DMSO-d6) δ:10.67(s,1H),7.53 (d,J=8.4 Hz,1H),7.31 (d,J= 1.6 Hz,1H),7.17 (d,J =2.2 Hz,1H),6.92 (dd,J =8.6,1.8 Hz,1H),5.36(d,J=4.5 Hz,1H),5.22(d,J=4.3 Hz,1H),5.02(d,J=5.6 Hz,1H),4.63(t,J=5.9 Hz,1H),4.59(d,J=7.2 Hz,1H),3.65 ~3.78(m,1H),3.55(dt,J=11.4,6.2 Hz,1H),3.22(tt,J=7.5,3.7 Hz,3H),3.12(td,J=9.1,8.7,4.8 Hz,1H);13C-NMR(125 MHz,DMSOd6)δ:137.8,133.8,126.7,119.5,119.0,112.7,111.6,104.7,77.7,77.1,73.9,70.6,61.5。
2.3 实验结果
2.3.1 化合物2 的合成影响因素
本研究对化合物(1-乙酰基-6-氯吲哚-3-基)-2,3,4,6-四乙酰基-β-D-葡萄糖苷的不同反应工艺条件进行了单因素优化,旨在得到最佳的合成条 件[11-12]。 具体反应式如图2 所示。
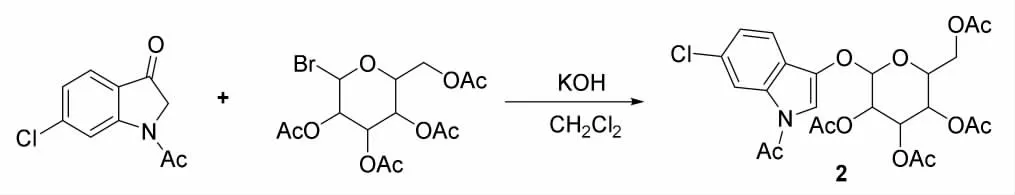
图2 化合物2 的合成路线
(1)反应物投料比对生成物收率的影响
设定NaNH2的加入量为1 mol, 反应物N-乙酰基-6-氯-3-吲哚酮与溴代四乙酰葡萄糖的物质的量比分别为1∶1,1∶2,1∶3,1∶4,在室温下反应12 h,考察2 种化合物的投料比对反应产物的影响。 结果见表1。 由表1 可知,当N-乙酰基-6-氯-3-吲哚酮的投料量恒定时, 增加化合物溴代四乙酰葡萄糖的量,能在一定程度上提高产物的收率;当两者的投料比达1∶3 时,产物的收率基本趋于稳定,反应过程接近平衡状态; 继续加入过多的溴代四乙酰葡萄糖,收率提高不明显,而且会造成原料的浪费,增加目标产物合成的成本。 因此选定反应物的投料比为1∶3。

表1 反应物投料比对生成物收率的影响
(2)NaNH2的量对生成物收率的影响
以N-乙酰基-6-氯-3-吲哚酮加入量1 mol 为基准, 反应物料N-乙酰基-6-氯-3-吲哚酮和溴代四乙酰葡萄糖的物质的量比为1∶3,N-乙酰基-6-氯-3-吲哚酮与NaNH2的物质的量比分别为1∶1,1∶2,1∶3,1∶4,在室温下反应12 h,考察NaNH2的量对生成物收率的影响。 结果见表2。 由表2 可知,适量地增加NaNH2的投料量,在一定程度上能促进反应的进行, 但是当NaNH2过量到一定程度时,NaNH2会影响溴代四乙酰葡萄糖的稳定性,进而影响生成物的收率。 因此确定反应中N-乙酰基-6-氯-3-吲哚酮与NaNH2的物质的量比为1∶2。
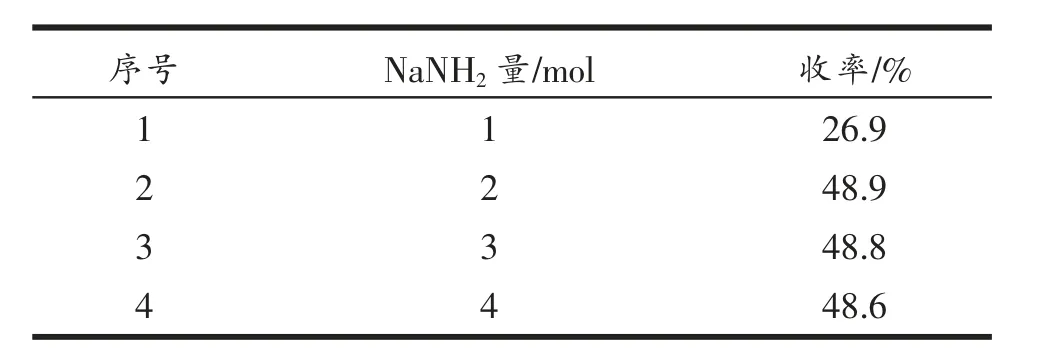
表2 NaNH2 量对生成物收率的影响
(3)反应时间对生成物收率的影响
设定反应物N-乙酰基-6-氯-3-吲哚酮与溴代四乙酰葡萄糖的物质的量比为1∶3、N-乙酰基-6-氯-3-吲哚酮与NaHN2的物质的量比为1∶2,在室温下分别反应8、10、12 和14 h, 探究反应时间对生成物收率的影响。 结果见表3。 由表3 可知,当反应进行12 h 时,产物收率最大。

表3 反应时间对生成物收率的影响
2.3.2 化合物1 的合成影响因素[12-13]
本实验中6-氯-3-吲哚-β-D-葡萄糖苷的合成路线的具体反应式如图3 所示。
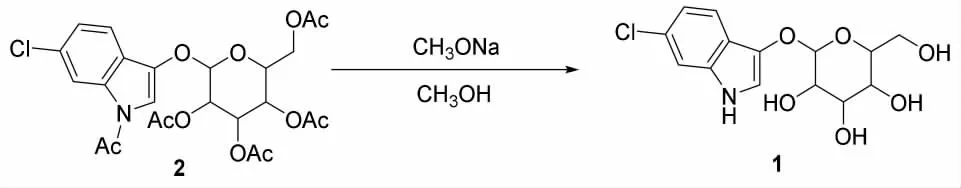
图3 化合物1 的合成路线
(1)不同碱对产物收率的影响
设定(1-乙酰基-6-氯吲哚-3-基)-2,3,4,6-四乙酰基-β-D-葡萄糖的投料量为1 mol, 不同碱(K2CO3、KOH、 NaOH 、NaNH2)的用量也为1 mol,在室温下反应4 h,考察不同碱对产物收率的影响。 结果见表4。 由表4 可知, 当反应过程中选取KOH 作为碱时,反应产物的收率最高。
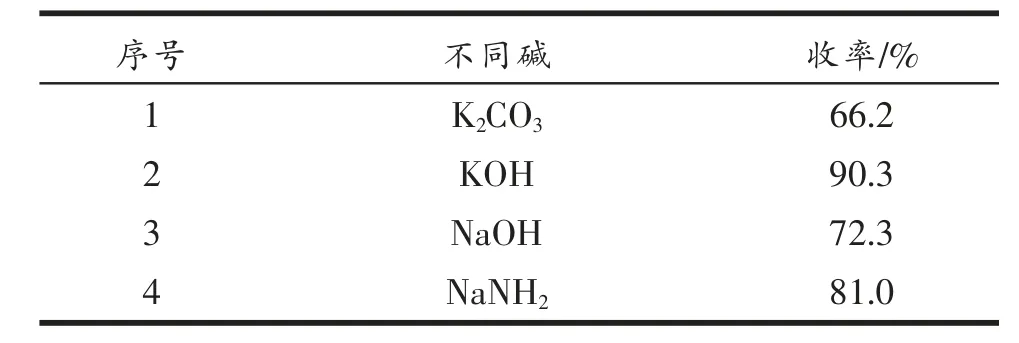
表4 不同碱对产物收率的影响
(2)KOH 的量对产物收率的影响
设定(1-乙酰基-6-氯吲哚-3-基)-2,3,4,6-四乙酰基-β-D-葡萄糖的投料量为1 mol,KOH 的用量分别为0.5、1.0、1.5 和2.0 mol,在室温下反应4 h,考察KOH 用量对产物收率的影响。结果见表5。由表5 可知,当(1-乙酰基-6-氯吲哚-3-基)-2,3,4,6-四乙酰基-β-D-葡萄糖与KOH 的投料比为1∶1 时, 反应产物的收率最大,增加碱的量,产物收率并未有明显变化。
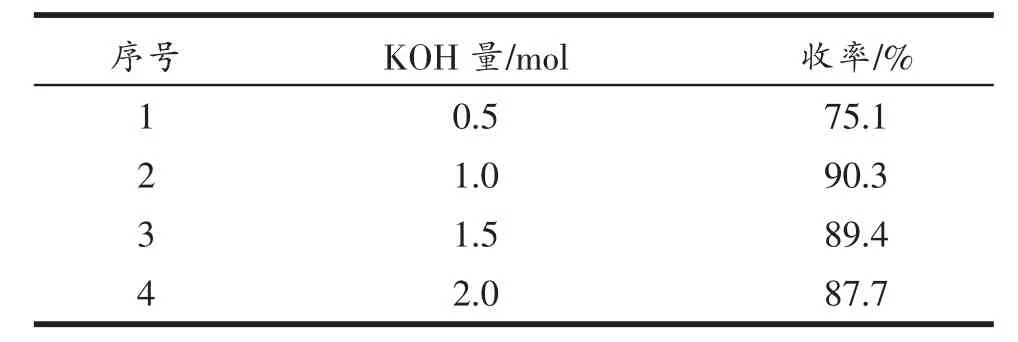
表5 KOH 用量对产物收率的影响
3 讨论与结论
近些年,显色培养基在食品行业、医药临床等病原微生物的检测领域应用非常广泛,它有效保证了病原微生物检测结果的准确性,同时又保证了检测过程的时效性, 简化了传统病原微生物检测过程[13-15]。吲哚酚类糖苷酶显色底物β-D-葡萄糖苷是病原微生物检测过程中常见的底物, 具有检测灵敏度高、操作简单等特点。
葡萄糖苷的构型主要分为α 与β 两种, 其中,β-D-葡萄糖苷具有动力学稳定构型[16],有文献报道[17-18],葡萄糖苷的合成产物中大多数都以α 与β的混合构型同时存在, 若能有效合成具有单一构型的β-葡萄糖苷,将在很大程度上提高病原微生物检测的准确性及灵敏度。
6-氯-3-吲哚-β-D-葡萄糖苷主要用于能够产生β-D-葡萄糖苷酶的病原微生物的检测,如大肠杆菌,大肠杆菌在自身代谢过程中产生β-D-葡萄糖苷酶,利用这一特性,检测过程中,将6-氯-3-吲哚-β-D-葡萄糖苷用作β-D-葡萄糖苷酶的检测底物,在其水解后, 有效解离出游离的色原6-氯-3-羟基吲哚,同时经过催化氧化反应后,产生了肉眼可见颜色的双吲哚产物, 进而确定β-D-葡萄糖苷酶的存在,最终检测出大肠杆菌病原微生物。
本文以试剂邻氨基苯甲酸作初始原料, 经过卤化、酰化、水解等反应,对吲哚酚类糖苷酶显色底物6-氯-3-吲哚-β-D-葡萄糖苷进行了全合成,经核磁氢谱对化合物结构进行表征, 分析了合成过程中关键的影响因素(反应时间、投料比及碱的用量等),最终确定了目标化合物的最佳合成路线。 此合成路线具有原料简单易得、 合成步骤较少及反应条件相对温和等特点, 为吲哚酚类糖苷酶显色底物的综合利用提供了技术支持。
