依普利酮合成工艺研究进展
2022-10-08于旭超叶有志蒋建武
于旭超,叶有志,蒋建武
(台州仙琚药业有限公司,浙江 台州 317016)
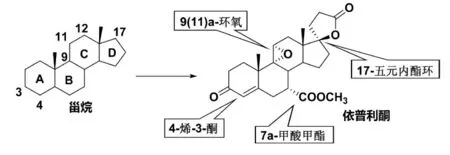
依普利酮(1)是一种选择性醛固酮受体拮抗剂,化学名为9,11α-乙氧基-17-羟基-3-氧-17α-孕-4-烯-7α,21-二羧酸-γ-内甲酯,分子式为C24H30O6,是由美国辉瑞公司开发并于2002 年批准上市的治疗高血压药物。分子结构见图1。

图1 依普利酮结构式
2003 年,美国食品药品管理局(FDA)批准依普利酮用于治疗急性心肌梗死导致的心衰。依普利酮特异性阻断醛固酮受体,而对雄激素和黄体酮受体的亲和力极低,性激素等相关不良反应少,耐受性好。对于联用多种降压药未能控制的重度高血压,依普利酮可起到明显的降压作用。对严重心力衰竭和心肌梗死病人,联用依普利酮可提高生活质量并降低死亡率。此外,依普利酮可以显著减轻肾小球的超滤作用,可减轻高血压患者的白蛋白尿,对于合并糖尿病的高血压患者,肾脏保护作用更为明显[1-3]。依普利酮作为重要的心血管疾病治疗药物,其合成工艺一直是制药领域的研究热点。本文综述现有的依普利酮合成方法,并展望未来工艺改进的方向。
1 合成方法
依普利酮的合成涉及甾烷4 个位点的修饰,即A 环上的α,β-不饱和酮,B 环上的α-甲酸甲酯,C 环上的α-环氧基和D 环上的β-羟基-内酯环(Scheme 1)。依普利酮的制备工艺通常采用半合成法,研究人员已发展了一系列以甾环化合物为原料合成依普利酮的方法。
Scheme 1
1.1 以坎利酮为原料合成依普利酮
Ng 等[4]报道了以坎利酮(canrenone)(2)为起始原料合成依普利酮的方法,主要涉及7α-甲酸甲酯的及9 (11)α-环氧基团的引入。首先通过生物发酵引入羟基,得到1lα-羟基坎利酮(3);在三乙基胺和氯化锂存在的条件下,3 与丙酮氰醇进行加成反应得到烯氨化合物4;在甲醇和水的溶液中,4 和盐酸反应得到双酮5,并经碱性条件下的开环酯化得到7α-甲酸甲酯化合物6;6 和甲磺酰氯、三乙胺进行甲磺酰化反应得到磺酸酯7,并经加热消除反应得到烯烃8;最后在三氯乙酰胺及磷酸氢二钾的存在下,8 和双氧水发生环氧化反应得到依普利酮(1)(Scheme 2)。

Scheme 2
随后,Ng 等[5]对上述合成工艺步骤顺序进行了优化。仍以坎利酮(2)为原料,先经相似的氰基加成、烯胺水解、二酮开环酯化得到7α-甲酸甲酯坎利酮(11),再经生物发酵引入羟基后得到化合物6,最终经消除及环氧化后得到目标产物依普利酮(1)(Scheme 3)。

Scheme 3
王少仲等[6]则直接以11α-羟基坎利酮(3)为起始原料,经丙酮氰醇加成、水解、开环酯化等反应得到7α-甲酸甲酯化合物6,6 在三氯氧磷和吡啶的存在下经一步反应消去11 位羟基,最后在双氧水的作用下环氧化得到依普利酮(1)(Scheme 4)。

Scheme 4
范丽丽等[7]以11α-羟基坎利酮(3)为原料,首先与2-甲基呋喃反应得到7α-甲基呋喃环-11α-羟基坎利酮化合物(12)。随后经磺酰化/消除两步反应得到9(11)-烯化合物14,再经氧化/酯化得到化合物8。8 在三氯乙酰胺和磷酸氢二钾的催化下,经双氧水环氧化得到目标产物依普利酮(1)。此路线用呋喃环作为引入7 位甲酸甲酯的预官能团,避免了使用剧毒的氰化物试剂,同时也避免了低温、臭氧等苛刻反应条件(Scheme 5)。

Scheme 5
化合物6 经一步法脱羟基或磺酰化/脱磺酸酯(两步法脱羟基)得到8 的过程中,会产生含Δ11(12)-双键的副产物。为了进一步提高脱水反应的选择性,谢祚宜等[8]对脱羟基工艺进行了改进。以磺酰酯化合物7 为原料,三氟乙酸为溶剂,在碱和醋酐的作用下,高效消除甲磺酸酯,从而得到Δ9(11)-烯酯化合物8,收率为90%左右,且反应生成的Δ11(12)-烯杂质含量较少(Scheme 6)。

Scheme 6
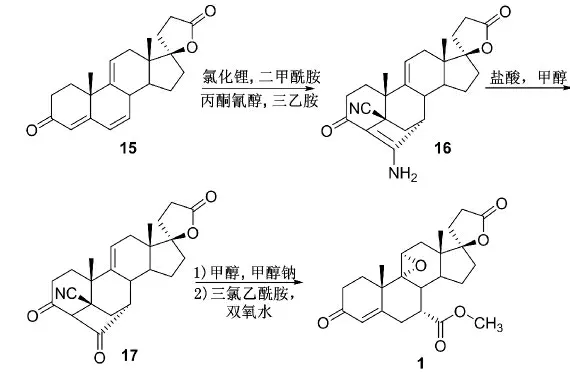
商艳梅等[9]则以Δ9(11)-坎利酮(15)为起始原料,再经相似的氰化物加成、烯胺水解、开环酯化、环氧化等过程制得产物依普利酮(1)。此合成路线较短,不存在传统脱水工艺中杂质多、反应不完全等诸多问题,有利于工业化生产(Scheme 7)。但该路线起始原料来源较少,成本较高。
Scheme 7
刘喜荣等[10]公开了一种依普利酮关键中间体化合物19 的制备方法。同样以Δ9 (11)-坎利酮(15)为起始原料,在铜盐的催化下,与烯基卤化镁偶联得到7α-乙烯基化合物18,在18 的溶液中通入氧气和臭氧的混合物进行氧化,再经过氧酸氧化后得到7α-甲酸化合物19。19 再经酯化、环氧化等过程得到目标产物依普利酮(Scheme 8)。

Scheme 8
1.2 以烯醇醚为原料合成依普利酮
Ng 等[4]公开了一种以烯醇醚20 为原料合成依普利酮的方法。首先经硫叶立德反应,20 转化成环氧化合物21。随后通过与丙二酸二乙酯/乙醇钠反应得到内酯化合物22。22 经脱羧反应后得到脱羧物23,最后经烯醚水解、溴代及消除等反应得到Δ9(11)-坎利酮(15)。Δ9(11)-坎利酮经与Scheme 7 相似的过程转化为依普利酮(1)(Scheme 9)。

Scheme 9
1.3 以去氢表雄酮为原料合成依普利酮
张映华等[11]报道了一种由去氢表雄酮(25)合成依普利酮中间体坎利酮(2)的方法。首先,去氢表雄酮通过沃式氧化转变为化合物26,随后3 位羰基经烯醇醚化保护得到烯醚27,再经与Scheme 9 相似的环氧开环/脱羧导入五元内酯环得到化合物28,并最终经四氯苯醌脱氢得到坎利酮。坎利酮经Scheme 2 或Scheme 3 的方法合成依普利酮(Scheme 10)。

Scheme 10
1.4 以氢化可的松为原料合成依普利酮
李金亮等[12]报道了以氢化可的松(29)为原料合成依普利酮的方法。首先氧化氢化可的松17位侧链,并对3 位酮基进行烯醚保护得到中间体30,再经四氯苯醌脱氢反应得到中间体31。31 经氰化、水解、酯化、脱氰基等步骤引入7α-甲酸甲酯得到32。32 经磺酰化/消去反应制得9,11-双键化合物33。最终,33 经环化、环氧开环、脱羧及环氧化等步骤制得依普利酮(1)(Scheme 11)。

Scheme 11
2 总结与展望
综上所述,研究人员已对依普利酮4 个修饰位点官能团的引入合成工艺进行详细的研究,并发展了一些行之有效的方法,但上述合成路线仍存在一定的问题:(1)涉及剧毒氰化物、重金属催化剂和昂贵试剂;(2)工艺路线长,某些步骤反应条件苛刻,操作复杂且收率不高,工业化应用难度较大。
随着人们对制药工艺安全、绿色的日益重视,以及医药产业的蓬勃发展,开发更为温和、高效、绿色的依普利酮合成工艺是未来制药工作者的研究重点。后续依普利酮制备工艺的改进策略主要包括:(1)相对于传统化学法,生物发酵具有选择性好,条件温和,步骤短等优势。在甲泼尼龙的制备工艺中,发酵法已成功应用于11 位羟基的引入,并通过后续官能团衍生化实现了环氧化物的制备。若生物法能够在6α-甲基的引入中获得突破,将极大缩短合成路线,避免剧毒氰化试剂的使用,简化工艺操作;(2)开发高选择性的11位脱水消除工艺。通过开发新型脱水试剂,实现由“两步”消除反应缩短至“一步”,提高工艺步骤的经济性,降低工艺成本。
