百香果高酯果胶-酚酸衍生物的分子特征、体外抗氧化和免疫活性
2022-09-30艾连中赖凤羲宋子波
高 凡,丁 宁,艾连中,赖凤羲,*,张 汇,宋子波
(1.上海理工大学健康科学与工程学院,上海食品微生物工程技术研究中心,上海 200093;2.云南猫哆哩集团食品有限责任公司,云南 玉溪 653100)
果胶为优良的亲水胶体,常作为胶凝剂、增稠剂、稳定剂、可食性薄膜、营养与药物载体等以及促进肠道健康的膳食纤维广泛用于食品中。商业上果胶主要源自柑橘皮、苹果渣和其他果皮,其主干结构为半乳糖醛酸(galacturonic acid,GalA)组成的同型半乳糖醛酸聚糖(homogalacturonan,HG),分枝结构主要为两种类型的鼠李半乳糖醛酸聚糖(rhamnogalacturonan,RG)(RG I和RG II),依GalA的羧基甲基酯化程度可分为高酯(酯化度(degree of esterification,DE)>50%)和低酯(DE<50%)果胶,各具有不同的流变特性与用途。
天然果胶的亲水性高,具有良好的凝胶性、增稠性和稳定性等重要的功能性,是优良的膳食纤维,可改善肠道菌相,但其抗氧化活性和低免疫调节活性相对较低,必须经酶解或酸热水解并进行分离和纯化等处理才能富集具有高免疫活性的RG I区域组分及大肠癌细胞高抑制活性的RG II区域组分。然而上述处理会使果胶分子降解,损失原来的功能特性。因此,采用无降解性方法修饰果胶的结构,以改良生物活性与疏水性相关的功能性是一重要的策略。
将膳食中常见的酚酸与果胶衍生化制备改性果胶是一种新趋势。在化学和生物衍生法中,利用漆酶催化酚酸接枝到果胶的方法备受瞩目,其具有天然、绿色安全、可回收性、操作简单及无需加热等优点,反应机理是漆酶将酚羟基氧化成中间产物-酚氧自由基,酚氧自由基进一步氧化断裂形成醌类物质,再亲核性作用于果胶分子GalA的自由羧基上形成酯基。初步被研究的果胶-酚酸衍生物有柑橘高酯果胶(citrus high-methoxy pectin,CHP)-阿魏酸(ferulic acid,FA)、柑橘低酯果胶(citrus low-methoxy pectin,CLP)-FA/咖啡酸(caffeic acid,CaA)与甜菜浆高酯果胶-FA等衍生物,这些衍生物具有较好的疏水性、乳化性与乳化稳定性、凝胶强度以及抗氧化活性(1,1-二苯基-2-三硝基苯肼(1,1-diphenyl-2-picrylhydrazyl,DPPH)自由基和2,2’-联氮-双-(3-乙基苯并噻唑啉-6-磺酸)(2,2’-azinobis(3-ethylbenzothiazoline-6-sulfonic acid),ABTS)阳离子自由基的清除能力)等。然而果胶结构及酚酸结构对其衍生物的体外抗氧化活性和免疫调节活性的影响鲜见研究。此外,对香豆酸(-coumaric acid,CA)和FA都属于羟基肉桂酸类,广泛存在于膳食水果和谷物中,均具有良好的抗氧化、抗菌、抗炎活性、抗糖尿病及免疫调节等作用。但相关研究大多以衍生FA和柑橘果胶为主,鲜有有关百香果果胶-CA衍生物的研究,且百香果果胶(新型果胶)具有与商业柑橘果胶相似的结构特征,可作为胶凝剂和稳定剂等加以应用,但目前研究仅限于其结构表征,其功能性亟待深入开发。
本研究旨在制备具有高体外抗氧化与免疫调节活性的果胶衍生物,以漆酶催化合成的百香果高酯果胶(passion fruit high-methoxy pectin,PFP)-CA和FA衍生物为主,并阐明不同衍生处理对产物的分子构型与体外生物活性的影响。所得结果同时与抗坏血酸(vitamin C,VC)衍生物(没有酚氧化成醌类并键结的作用)和只添加漆酶不加酚酸的对照组作对比,以厘清酚酸组成对衍生物的分子构型与体外生物活性的影响与反应机理。本研究成果可为改性果胶在药食同源食品中的应用提供理论依据。
1 材料与方法
1.1 材料与试剂
紫色百香果(Sims f.)果皮由云南猫哆哩集团食品有限责任公司提供;小鼠巨噬细胞RAW264.7购自中国科学院上海生命科学研究院细胞资源中心。
漆酶(酶活力0.84 U/mg) 美国Sigma-Aldrich公司;无水乙醇 上海泰坦科技股份有限公司;DPPH、FA、CA、VC、硫酸亚铁 上海源叶生物科技有限公司;溴化钾(光谱级) 上海阿拉丁生化科技股份有限公司;硝酸钠、过氧化氢 国药集团化学试剂有限公司;一氧化氮检测试剂盒、中性红细胞增殖及细胞毒性检测试剂盒、噻唑蓝(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide,MTT)细胞增殖及细胞毒性检测试剂盒 上海碧云天生物技术有限公司;胎牛血清、青霉素-链霉素双抗 维森特生物技术(南京)有限公司;DMEM培养基 美国Gibco公司。
1.2 仪器与设备
1260高效分子筛色谱-多角度激光光散射(high performance-size exclusion chromatography-multiangle laser light scattering,HPSEC-MALLS)仪 美国Wyatt Technology公司;Nicolet 380傅里叶变换红外光谱仪美国Thermo Fisher Scientific公司;725型紫外-分光光度计上海光谱仪器有限公司;RCT基本型(安全控制型)磁力搅拌器 德国IKA公司;SU8010场发射扫描式电子显微镜 日本Hitachi公司;SpectraMax i3酶标仪美国Molecular Devices公司;SW-CJ-FD超净工作台苏州净化设备有限公司;Ardy Bio微孔板振荡器 梅洁(上海)科技有限公司。
1.3 方法
1.3.1 百香果果胶的提取
PFP提取方法参照丁宁等的方法,提取试剂为酸化水溶液(用0.1 mol/L HNO溶液调节至pH 2.1)、料液比1∶20(/)、85 ℃加热3 h。粗提取液经离心(6 000 r/min、20 min)得上清液,真空浓缩,以2 倍体积的体积分数95%乙醇溶液沉淀果胶、离心(6 000 r/min、20 min)收集胶体、冷冻干燥,得到PFP,其组成中半乳糖醛酸物质的量分数为78.5%,DE为75.2%,所用PFP的糖类组成与分子结构特征与商业CHP极相似。
1.3.2 百香果果胶的改性
果胶与酚酸反应的漆酶作用条件参考Karaki等的方法并稍作修改,取1 g PFP溶于90 mL磷酸盐缓冲液(50 mmol/L、pH 6.5)中,在80 ℃下磁力搅拌1 h,另将固定量的酚酸(0.18 g FA和0.21 g CA,相当于果胶(游离羧基)∶(酚酸)=1∶1)预溶解于10 mL纯甲醇中,将上述两种溶液在30 ℃下混合后,添加漆酶10 U(11.91 mg)反应4 h,包裹锡箔纸在冰箱4 ℃暗室下反应24 h,在蒸馏水中透析(8~14 kDa)24 h,每隔6~8 h换一次水,以除去未衍生的酸,旋转蒸发浓缩后冷冻干燥得到百香果高酯果胶-阿魏酸衍生物(PFP-laccase-FA,PFP-L-FA)和百香果高酯果胶-对香豆酸衍生物(PFP-laccase-CA,PFP-L-CA)。另制备只加漆酶的高酯果胶衍生物(PFP-laccase,PFP-L)以及高酯果胶-VC衍生物(PFP-laccase-VC,PFP-L-VC)作为对照组,其中VC的反应量为0.19 g。反应酸的用量根据:1 g PFP(脱水半乳糖醛酸相对分子质量176)的游离羧基物质的量为0.001 1 mol;故(游离羧基)∶(酸)=1∶1下所需的酸为0.181 g FA、0.214 g CA及0.194 g VC(FA相对分子质量194.2、CA相对分子质量164.2、VC相对分子质量176.1)。
1.3.3 总酚含量的测定
参照Sato等的方法测定总酚含量。以没食子酸为标准品,线性回归方程为=0.033 8-0.029 9(=0.992),其中为720 nm波长处的吸收度,为没食子酸质量浓度/(μg/mL)。样品中总酚含量(以没食子酸当量计)计算如公式(1)所示。
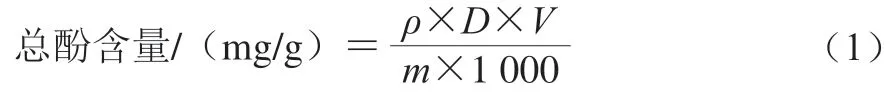
式中:为样品液中多酚的质量浓度/(μg/mL);为稀释倍数;为样品液总体积/mL;为样品质量/g。
1.3.4 分子质量相关参数的测定
用流动相(0.1 mol/L NaNO和质量分数0.02%NaN)溶解制备质量浓度1 mg/mL PFP及其衍生物溶液,0.22 μm铝膜过滤后取100 μL注入HPSEC系统分析,该系统配备MALLS检测器和示差折射(differential refractive index detector,DRI)检测器,串联一支保护管柱和两支分析管柱,分别为ShodexOHpak SB-G 6B(500 mm×6 mm,10 μm)、SB-805 HQ(300 mm×8 mm,13 μm)和SB-803 HQ(300 mm×8 mm,6 μm),在40 ℃下以流速0.6 mL/min洗脱80 min;折射率增量(d/d)设置为0.145 mL/g,并采用ASTRA 7.1.3软件进行数据采集、处理和分析。
果胶大分子的构象表征通过Mark-Houwink-Sakurada(MHS)方程(式(2))和非均向性构型参数=/来表示,与分子链构象的刚性和硬度呈正比,指数<0.775、1.5~1.8和>2分别表示构象为紧密的球状、柔软的无规则线状和伸展性分子,其中和分别通过Flory-Fox公式(式(3))和Einstein-Simha公式(式(4))计算得出。


式中:[]为特性黏度/(dL/g);为聚合物-溶剂交互作用参数/(mL/g);为果胶的摩尔质量/(g/mol);为构形参数;与均与聚合物构象有关,指数>1.4、0.8~1.4、0.5~0.8和0.2~0.5和0.0时,分子构象分别为硬杆状、刚性杆状、半柔性线圈、无规则线圈和球状球体或高度支链;为环动半径/nm;是Flory-Fox参数(约为2.6×10kg,无规则线圈状聚合物分子),与链的刚性有关;为水合动态半径/nm;是阿伏伽德罗常数(6.02×10mol)。
1.3.5 傅里叶变换红外光谱测定
采用KBr压片法,分别将2 mg冻干果胶及其衍生物粉与100 mg KBr置于玛瑙研钵中研磨压片制成直径7 mm的薄片,使用傅里叶变换红外光谱仪对其进行测定,以空气作为背景,扫描范围4 000~400 cm,扫描次数64 次,分辨率16 cm,使用Omnic软件进行数据采集和分析。
1.3.6 微观结构观察
将PFP和4 种衍生物磨成粉末状,然后取适量粉末置于导电碳胶带上,喷金处理,冷却至常温后用电子显微镜扫描,在放大100 倍和1 000 倍下观察果胶及其衍生物的微观结构。
1.3.7 DPPH自由基清除率测定
DPPH自由基清除率测定参照Brand-Williams等的实验方法稍作修改。将2.0 mL新鲜配制的DPPH溶液分别与2.0 mL不同质量浓度的果胶及其衍生物溶液(质量浓度0~2 mg/mL)充分混匀,室温避光反应30 min后,以体积分数80%乙醇溶液调零,在517 nm波长处测定吸光度,以VC为阳性对照,按照公式(5)计算DPPH自由基清除率。
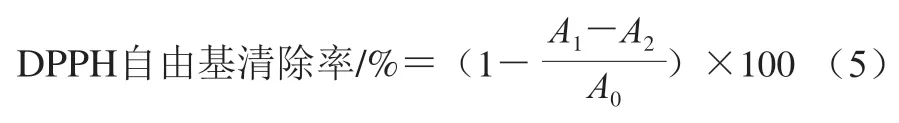
式中:为空白对照(体积分数80%乙醇溶液代替样品或VC)的吸光度;为DPPH溶液与样品或阳性对照VC的吸光度;为体积分数80%乙醇溶液代替DPPH溶液的吸光度。
1.3.8 超氧阴离子自由基清除率测定
超氧阴离子自由基清除率测定参照Fridovich的实验方法稍作修改。将1.0 mL样品溶液(质量浓度0~2 mg/mL)、1.0 mL 108 μmol/L氯化硝基四氮唑蓝和1.0 mL 468 μmol/L烟酰胺腺嘌呤二核苷酸溶液混合,混合均匀后加入1.0 mL 60 μmol/L吩嗪硫酸甲酯溶液,将上述混合好的溶液室温下静置5 min,在560 nm波长处测定吸光度,以上溶液均用Tris-HCl缓冲液(16 mmol/L、pH 8.0)配制,以VC为阳性对照,按照公式(6)计算超氧阴离子自由基清除率。
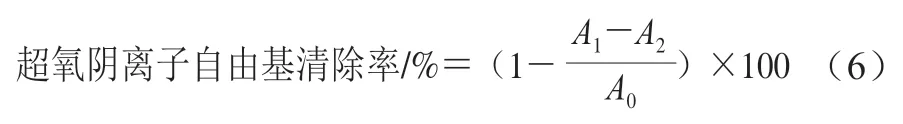
式中:为空白对照(Tris-HCl缓冲液代替样品或VC)的吸光度;为显色体系与样品或阳性对照VC的吸光度;为缓冲液与样品或VC的吸光度。
1.3.9 羟自由基清除率测定
羟自由基清除率测定参照Smirnoff等的实验方法稍作修改。将0.6 mL 6 mmol/L FeSO·7HO溶液与2.0 mL果胶及其衍生物溶液(质量浓度0~2 mg/mL)分别混匀后,加入0.6 mL 6 mmol/L的HO溶液,充分混匀后反应10 min,再向各管加入0.6 mL 6 mmol/L水杨酸-无水乙醇溶液,将各管中的溶液充分混合反应10 min,在510 nm波长处测定吸光度,以VC为阳性对照,按公式(7)计算羟自由基清除率。

式中:为空白对照(蒸馏水代替样品或VC)的吸光度;为显色体系与样品或阳性对照VC的吸光度;为蒸馏水代替HO溶液的吸光度。
1.3.10 Fe螯合率测定
Fe螯合率测定参照Dinis等的实验方法稍作修改。将1 mL不同质量浓度(0~2 mg/mL)的果胶及其衍生物溶液和0.1 mL 2 mmol/L FeCl溶液、0.2 mL 5 mmol/L啡啰嗪-钠盐溶液混合,用3.7 mL蒸馏水补至5 mL混匀。将上述混合好的溶液在室温条件下进行孵育10 min,在560 nm波长处测定吸光度。空白对照组为蒸馏水,乙二胺四乙酸二钠(ethylene diamine tetra-acetic acid disodium salt,EDTA-2Na)为阳性对照,按公式(8)计算Fe螯合率。
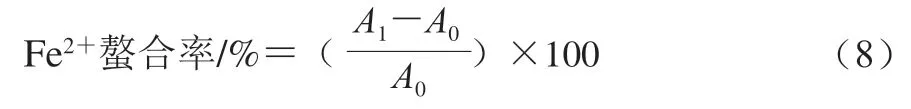
式中:为空白对照(蒸馏水代替样品或EDTA-2Na)的吸光度;为样品与显色体系的吸光度。
1.3.11 总还原力测定
总还原力测定参照Benzie等的方法稍作修改。取2 mL不同质量浓度(0.1~2.0 mg/mL)的样品,加入pH 6.6的0.2 mol/L磷酸缓冲液和质量分数1%铁氰化钾溶液各2 mL,混匀,50 ℃水浴反应20 min,然后迅速冷却,添加2 mL质量分数10%的三氯乙酸溶液,然后将上述的混合液振荡使其均匀,3 000 r/min离心10 min,取上清液2 mL,加入2 mL去离子水和1 mL质量分数0.1%的三氯化铁溶液,静置10 min后在700 nm波长处测定吸光度,吸光度的大小直接反映还原性的强弱,以VC作阳性对照。
1.3.12 小鼠巨噬细胞RAW264.7培养
将小鼠巨噬细胞RAW264.7培养在含有10%胎牛血清、100 U/mL青霉素和质量浓度100 g/mL链霉素的DMEM培养基中,放置于37 ℃、含有5% CO和95%空气的饱和湿度培养箱中培养,培养约24 h(传代一次),以保持细胞处在对数生长期。
1.3.13 小鼠腹腔巨噬细胞RAW264.7增殖活性的测定
采用MTT法分析百香果果胶及其衍生物对小鼠腹腔巨噬细胞RAW264.7活力的影响。取对数生长期的细胞以2×10个/mL接种于96 孔板中,细胞培养24 h后,移去上清液,加入100 μL不同质量浓度(0.2~1.0 mg/mL)的果胶、果胶衍生物溶液或100 μL 0.001 mg/mL脂多糖(lipopolysaccharide,LPS)溶液(阳性对照),将小鼠腹腔巨噬细胞RAW264.7培养24 h,然后向每孔加入20 μL质量浓度5 mg/mL MTT溶液,经过孵育4 h后,将96 孔板中所有的液体移除,然后使用冷的磷酸盐缓冲液(0.01 mol/L、pH 7.4)清洗2 次,之后向每孔中加入150 μL二甲基亚砜溶剂,在微孔板振荡器上振荡混匀持续10 min,酶标仪测定570 nm波长处的吸光度。按公式(9)计算细胞存活率。
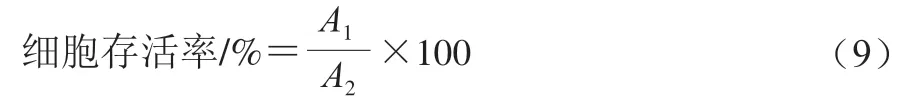
式中:为果胶及其衍生物在570 nm波长处的吸光度;为空白组(磷酸盐缓冲液)在570 nm波长处的吸光度。
1.3.14 小鼠腹腔巨噬细胞RAW264.7 NO分泌量的测定
采用Griess法测定果胶及其衍生物对巨噬细胞RAW264.7NO生成量的影响。取对数生长期的细胞以2×10个/mL接种于96 孔板中,细胞培养24 h后,吸取50 μL上清液加入至新的96 孔板中,加入100 μL不同质量浓度(0.2~1.0 mg/mL)的果胶、果胶衍生物溶液或100 μL 0.001 mg/mL LPS溶液(阳性对照),空白组为0.01 mol/L磷酸盐缓冲液代替果胶溶液样品,在室温下向96 孔板中分别加入50 μL的Griess试剂I和试剂II,缓慢混合均匀,用酶标仪测定540 nm波长处的吸光度。根据试剂盒的使用方法,绘制NO浓度标准曲线(=0.005 5+0.056 7,=0.999 2,其中为,为NO浓度/(μmol/L),测定NO浓度范围0~100 μmol/L),并计算NO的浓度。
1.3.15 小鼠腹腔巨噬细胞RAW264.7吞噬能力的测定
采用中性红吞噬实验法来评价果胶及其衍生物对小鼠腹腔巨噬细胞吞噬能力的影响。取对数生长期的细胞以2×10个/mL接种于96 孔板中,将巨噬细胞RAW264.7培养24 h,去除96 孔板中的上清液,加入100 μL不同质量浓度(0.2~1.0 mg/mL)的果胶、果胶衍生物溶液或100 μL质量浓度0.001 mg/mL LPS溶液(阳性对照),细胞培养24 h后,之后向96 孔板中分别加入100 μL质量浓度1 mg/mL中性红生理盐水,将上述混合液孵育30 min,然后将96 孔板中的上清液去除,为了去除残余的中性红试剂,需要用冷的0.01 mol/L磷酸盐缓冲液清洗3 遍,然后向96 孔板中分别加入100 μL的细胞裂解液,将混合的溶液室温下静置2 h,等到96 孔板中的细胞溶解之后,利用酶标仪测定540 nm波长处的吸光度。按公式(10)计算吞噬能力。
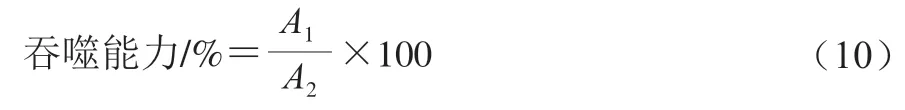
式中:为果胶及其衍生物的吸光度;为空白(磷酸盐缓冲液)的吸光度。
1.4 数据处理与分析
所有实验均重复3 次,数据以平均值±标准差表示。显著性差异分析用SPSS Statistics 24.0软件通过Duncan法进行多重比较确定,<0.05时表示具有显著性差异。用OriginPro 2021b软件绘图。
2 结果与分析
2.1 果胶提取物PFP及其衍生物的总酚含量分析
表1显示PFP的总酚含量为3.66 mg/g,只加漆酶的样品PFP-L总酚含量显著降至2.38 mg/g(<0.05),可能因为漆酶是多酚氧化酶,与酚酸作用后,酚酸被氧化为酚氧自由基,进而氧化形成半醌类物质,因此PFP的总酚含量显著降低。PFP-酚酸衍生物中,PFP-L-CA的总酚含量最高(12.29 mg/g)(衍生率约5.8%),其次为PFP-LFA(11.07 mg/g)(衍生率约6.2%),显著高于PFP-L-VC(4.68 mg/g)、PFP和PFP-L(<0.05)。故可推知有大量的FA和CA存在于PFP衍生物上。PFP-L-FA和PFP-L-CA的总酚含量略高于柑橘低酯果胶-咖啡酸交联物(citrus low-methoxyl pectin-caffeic acid conjugate,CLP-CaA)的总酚含量(9.6 mg/g)。PFP的总酚含量略高于低温(50 ℃)水萃取的百香果高酯果胶(DE 57%)的总酚含量(0.71 mg/g)和低酯/高酯柑橘果胶(分别为1.20 mg/g和1.77 mg/g),但低于酸提与汽爆提取的百香果高酯果胶(分别为8.96 mg/g和18.82 mg/g),这可能与果胶的提取方式、DE和种类不同相关。
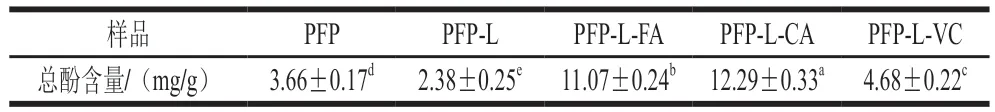
表1 果胶提取物PFP及其衍生物的总酚含量Table 1 Total phenolic contents of PFP and its derivatives
2.2 果胶及其衍生物的分子质量及构象表征分析
图1为果胶及其衍生物(PFP-L、PFP-L-FA、PFP-L-CA和PFP-L-VC)在0.1 mol/L硝酸钠溶液中分子质量分布的HPSEC-MALLS色谱图。图1A显示PFP的分子分布,其中折射指数(refractive index,RI)信号(信号幅度与样品浓度成正比)显示单一分子分布,出峰时间在19~28 min之间,而光散射(light scattering,LS)信号(与分子粒径和浓度成正比)呈双峰(20.5 min和24 min),显示有两种分子构型密度或粒径存在。只添加漆酶的PFP-L(图1B)的果胶分子的主峰尖峰时间(24 min)与PFP相似,但大粒径尖峰(LS在20.5min处的肩峰)信号幅值减小,26 min处增加的小粒子肩峰表明PFP分子粒径或构型被部分修饰,而35 min处小尖峰为漆酶的信号峰。PFP-L-FA(图1C)和PFP-L-CA(图1D)皆呈现果胶分子LS信号的主尖峰尖锐化且尖峰时间前移到20.5 min,其中PFP-L-FA的大粒径信号(18 min)幅值减小,明显增加了小粒径肩峰(24 min)。PFP-L-VC(图1E)主尖峰尖锐化且整体出峰时间延后(即分子质量和粒径减小)且出现新的小粒径区域组分(33 min),显示果胶分子部分水解。
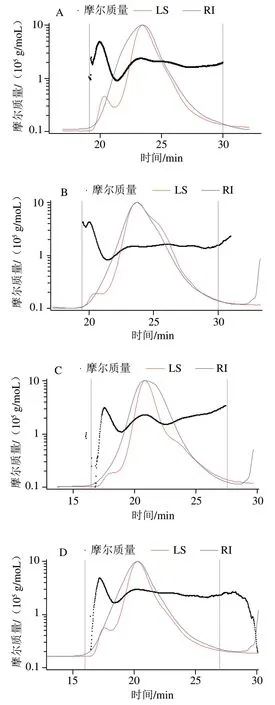
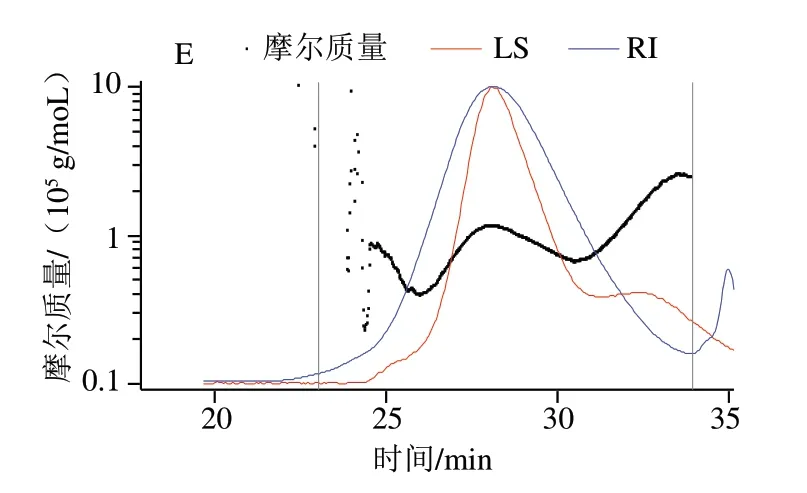
图1 PFP(A)、PFP-L(B)、PFP-L-FA(C)、PFP-L-CA(D)和PFP-L-VC(E)分子分布的HPSEC色谱图Fig. 1 HPSEC chromatograms for molecular mass distributions of PFP (A), PFP-L (B), PFP-L-FA (C), PFP-L-CA (D), and PFP-L-VC (E)
表2显示PFP及其衍生物的分子质量参数,PFP的平均重均分子质量()为190.5 kDa,平均数均分子质量()为175.1 kDa,平均多分散性指数(polydispersity index,PDI)(PDI=/)为1.09,固有黏度[]平均为6.09 dL/g,MHS关系式的参数=0.017 mL/g、=0.875;而假设果胶分子在0.1 mol/L硝酸钠溶液中为球状,将上述和[]值代入Flory-Fox和Einstein-Simha公式,所得平均环动半径为44.5 nm、平均水合动态半径为25.62 nm、非均向性构型参数为1.74。与PFP比较,PFP-L的、、PDI、[]、和均明显降低,构型参数和值略降低但仍接近PFP的构型参数;PFP-L-FA的和变化不明显,明显降低至19.52 nm,但PFP-L-CA的和均明显增加,明显升高至25.95 nm,两者的PDI、[]、值、以及值均明显降低,分别降至1.06和1.03、2.77 dL/g和4.65 dL/g、0.393和0.265、28.3 nm和34.0 nm及1.45和1.31。PLP-L-VC的和降至不到PFP的一半,伴随[]、值、以及大幅降低,分别降至0.97 dL/g、0.290、27.4 nm和10.84 nm;而PDI和值明显增加,分别为1.14和2.53。整体上,MHS参数的值随值降低而增加。
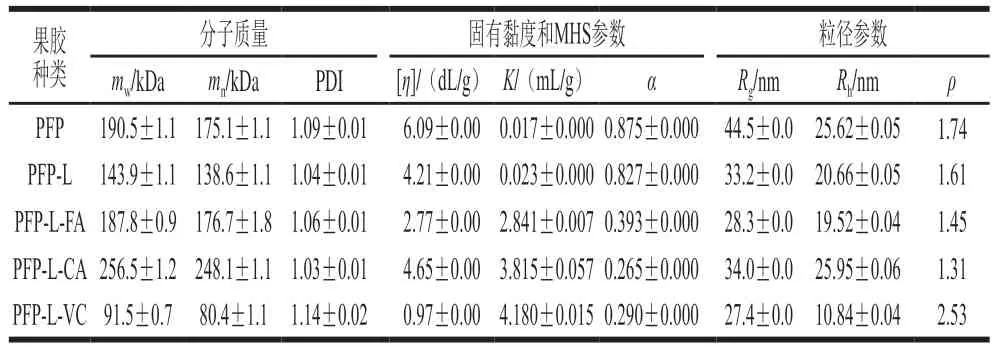
表2 果胶提取物PFP及其衍生物的分子链特征Table 2 Molecular chain characteristics of PFP and its derivatives
综上,CA衍生会促使PFP果胶的分子质量增加,而FA衍生对分子质量无明显影响,两种果胶衍生物的分子粒径相关的参数([]、和值)与构型参数(值和值)均明显降低。在0.1 mol/L的硝酸钠溶液中,PFP和PFP-L(值分别为0.875和0.827)呈刚性短杆状(值0.8~1.4),但PFP的环动半径和非均向性构型参数值较大,显示链段较硬些。PFP-L-FA和PFP-L-CA衍生物(值分别为0.393和0.265)呈无规则线圈状构型(值0.2~0.5),其值<1.5显示分子构型均向性较高、偏向柔软的无规则线状链。PFP-L-VC的分子质量与分子粒径相关的参数大幅降低,但值大幅增加到2.53,表明链段比PFP更刚硬,可能是VC导致果胶分子(尤其是中性糖分支)部分水解后,伸展性较高的HG链段占比增加所致。
与商业CLP(=226 kDa、PDI=2.62、[]=2.23 d L/g、=0.436、=0.552、=38.9 nm、=16.35 nm、=2.38)相比较,本研究PFP的、PDI、和值较低,而[]、值、及较高,显示PFP比CLP有较均一的分子质量分布、较大的分子粒径、较刚硬又较均向的分子链段。PFP-L-CA与同样是漆酶催化的CLP-CaA(=271 kDa、PDI=2.38、[]=1.83 dL/g、=0.479、=0.524、=30.6 nm、=16.24 nm、=1.88)发生的分子性质变化极为相似,均为分子质量与增加,伴随PDI、[]、值、和值减小,且无明显变化。不同的是,PFP-L-Ca的和值的变化程度相当大,但CLP-CaA变化程度相当小。综上,酚酸(FA、CA、CaA)衍生或不影响或增加果胶衍生物分子质量,但会修饰衍生物的分子构型使分子体积/粒径减小、链段较柔软、分子构型呈更紧密的无规则卷曲线圈状。漆酶催化合成果胶-酚酸衍生物不会发生降解现象,增稠和凝胶特性优于VC衍生物和化学衍生的阿拉伯木聚糖-儿茶素,后两者在衍生后分子质量降低,会损失增稠和凝胶特性。
2.3 傅里叶变换红外光谱分析
图2为衍生前后果胶的傅里叶变换红外光谱图,果胶PFP在3 446 cm处附近的宽峰为O-H的伸缩振动所引起,为糖类的特征吸收峰。在2 932 cm处为多糖C-H键的伸缩振动所引起;1 747 cm和1 641 cm处的伸缩振动分别对应于甲酯化和游离羧基中的C=O键的拉伸振动信号;1 437 cm处是COO的不对称伸缩振动吸收峰;1 385 cm和1 242 cm处是C-H伸缩振动吸收峰;1 110 cm和1 023 cm处的吸收峰与糖环和侧链上的C-C-O拉伸振动有关,为GalA的特征信号;1 200~800 cm波数之间的光谱被认为是碳水化合物的“指纹”区域。和PFP相比,PFP-L的吸收峰位置和峰面积无明显变化;然而衍生物在1 625 cm附近处的峰面积明显减小,1 739 cm附近处的峰面积明显增加,此结果表明衍生物发生了羧基酯化现象,类似CLP-FA或CLP-CaA、白萝卜果胶-FA以及羧基化可得然胶-FA衍生物的情形,其特征峰变化在1 519 cm或1 518 cm(CaA或FA)和1 616 cm或1 732 cm(分别为自由或酯化羧基)处,表明酚酸极可能通过酯键结合到果胶的羧基上。PFP-L-VC为无酚氧化成醌类并键结的参考组,图谱与PFP-L相似。
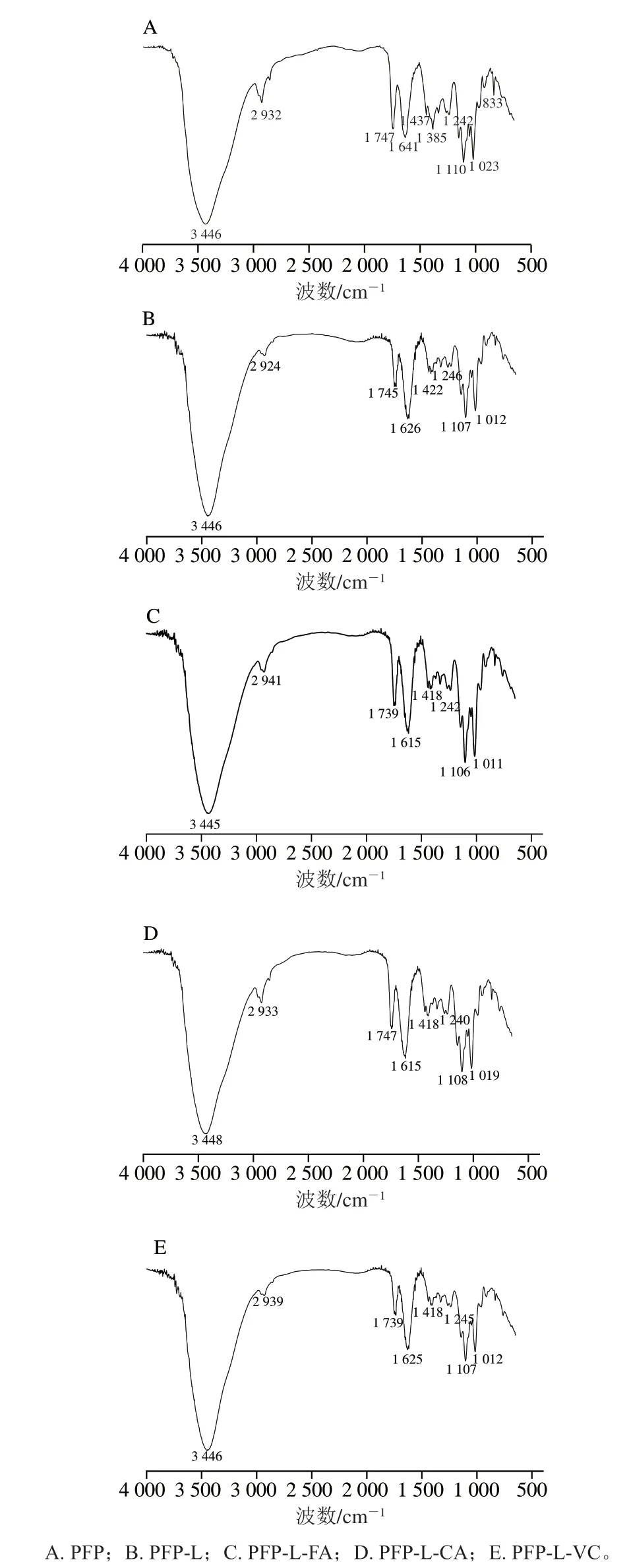
图2 果胶提取物PFP及其衍生物的傅里叶变换红外光谱图Fig. 2 Fourier transform infrared spectra of PFP and its derivatives
2.4 果胶提取物PFP及其衍生物的微观形态分析
图3显示果胶PFP及其衍生物的干燥粉粒的微观形态。PFP在放大100 倍下(图3A)呈现不规则形状的碎片与颗粒,在放大1 000 倍下(图3A)清楚可见这些碎片黏附紧密。PFP-L在放大100 倍下(图3B)表面呈现出松散的不规则絮状碎片,在放大1 000 倍下(图3B)碎片显得松散且部分卷曲。PFP-L-FA在放大100 倍下(图3C)呈光滑的大片折叠、松散并稍微卷曲,放大1 000 倍下(图3C)片状非常光滑。PFP-L-CA(图3D)和PFP-L-VC(图3E)在放大100 倍下都呈现不规则长条的絮状片,介于PFP-L和PFP-L-FA的中间混合形态。以放大1 000 倍观之,PFP-L-CA(图3D)的絮状片呈不规则的断层状,而PFP-L-VC(图3E)絮片呈大片折叠且平整光滑。显然,百香果皮果胶经过添加漆酶和酚酸衍生和VC作用后,粉粒的微观结构会明显改变,呈松散且大片絮状化。此现象与CLP-CaA干燥膜的结构呈絮状片且比CLP松散的现象一致。

图3 果胶提取物PFP及其衍生物的微观形态Fig. 3 Scanning electron micrographs of PFP and its derivatives
2.5 果胶提取物PFP及其衍生物的体外抗氧化活性分析
图4显示PFP及其酚酸衍生物对DPPH自由基、羟自由基、超氧阴离子自由基清除率以及Fe螯合率和总还原力均呈明显的剂量-效应关系。图4A说明质量浓度0.1~2.0 mg/mL的PFP对DPPH自由基的清除率达到18%~27%,与PFP-L接近。质量浓度1.5~2.0 mg/mL的PFP-L-CA DPPH自由基清除率高达65%~70%,约达到VC的70%(阳性对照)。然而,与PFP相比,PFP-L-FA和PFP-L-VC在所研究的质量浓度范围内DPPH自由基清除率明显较低。对于超氧阴离子自由基的清除率(图4B),PFP-L-CA和PFP-L-FA的清除率随着质量浓度(1~2 mg/mL)增加而升高,与PFP相比明显升高,在质量浓度2.0 mg/mL时均达到100%,与VC的清除率接近。质量浓度0.1~2.0 mg/mL PFP的超氧阴离子自由基清除率(40%~50%)在所有样品中处于中等水平,且PFP-L和PFP-L-VC的超氧阴离子自由基清除率随着质量浓度的提高平稳增加,在质量浓度2.0 mg/mL时达到约50%,与PFP相同。超氧阴离子自由基清除率对所有样品都表现出明确的浓度依赖性。对于羟自由基的清除能力(图4C),所有改性果胶,尤其是PFP-L-FA,在质量浓度0.1~2.0 mg/mL时清除率为40%~78%,明显高于PFP(15%~40%),且呈样品浓度依赖性。与PFP相比,所有改性果胶都显示出Fe螯合能力(图4D)和还原能力(图4E)增强。
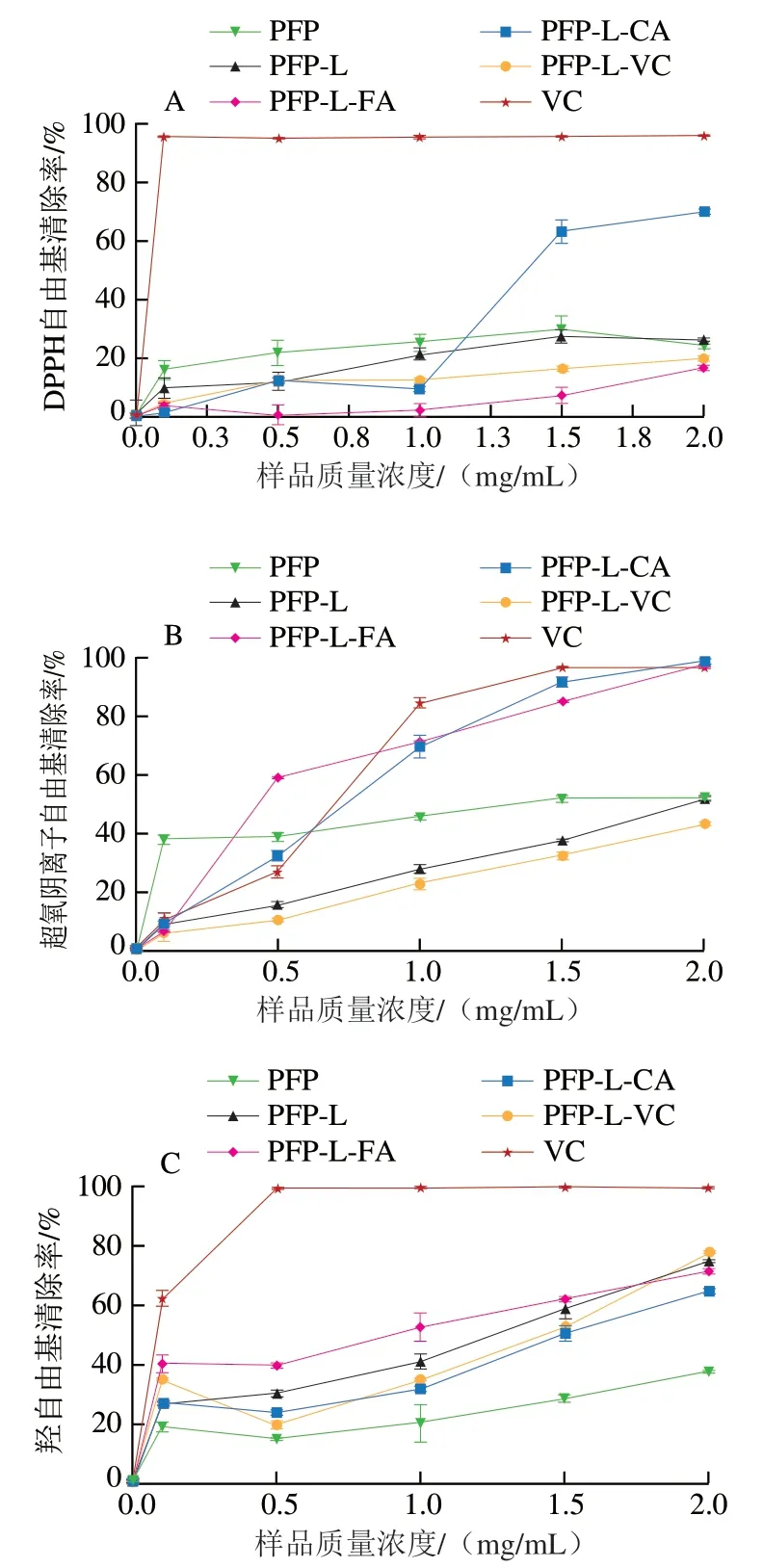
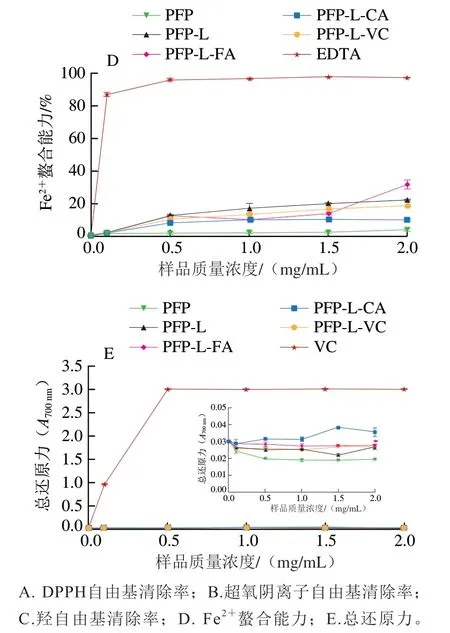
图4 果胶提取物PFP及其酚酸衍生物的抗氧化活性Fig. 4 Antioxidant activities of PFP and its derivatives with phenolic acids
本研究发现CA和FA的衍生作用可赋予果胶更强的体外抗氧化活性,能明显提高超氧阴离子自由基和羟自由基的清除率。以半抑制浓度(50% inhibiting concentration,IC)评价抗氧化活性,从DPPH自由基清除率方面考量,PFP-L-CA的抗氧化活性(IC=1.4 mg/mL)高于PFP-L-FA,略高于化学衍生的CHP-儿茶素或芦丁衍生物(IC=1.6~1.8 mg/mL),与果胶-FA衍生物及羧基可得然胶-FA衍生物(两者IC均为1.4 mg/mL)相同,略低于CLP-CaA和化学衍生的CHP-槲皮素衍生物(两者均IC=0.8 mg/mL)。对超氧阴离子自由基清除率的抗氧化活性方面,PFP-L-FA抗氧化活性(IC=0.45 mg/mL)高于PFP-L-CA、CLP-CaA与化学法衍生得到的壳聚糖-FA(综合IC=0.7~0.8 mg/mL)。在羟自由基清除率的抗氧化活性方面,PFP-L-FA的抗氧化活性(IC=1.0 mg/mL)高于PFPL-CA(IC=1.5 mg/mL),略低于CLP-CaA(IC=0.92 mg/mL),低于壳聚糖-FA衍生物(IC=0.76 mg/mL)。PFP-L-FA和PFP-L-CA的Fe螯合率与还原力虽不高,但仍优于PFP,与CLP-CaA的相似。
比较本研究和高凡等的研究结果发现,天然低酯果胶CLP羟自由基清除力(IC=2.0 mg/mL)优于对DPPH自由基和超氧阴离子自由基的清除力,且高于天然高酯果胶PFP,这可能与CLP含游离羧基较多有关。酚酸衍生处理明显提高果胶衍生物的抗氧化活性,且CLP衍生物提升效果优于PFP衍生物,超氧阴离子自由基清除率>羟自由基清除率;对醇溶体系的DPPH自由基清除力的促进效果为PFP-L-CA和CLP-CaA作用明显,而PFP-L-FA作用不明显;但FA衍生对水溶液体系的超氧阴离子自由基和羟自由基清除力的抗氧化活性促进效果优于CA衍生。由于总酚含量在上述样品中相差不多(PFP-L-FA总酚含量11.07 mg/g、PFP-L-CA总酚含量12.29 mg/g、CLP-CaA总酚含量9.6 mg/g),说明酚酸结构对抗氧化活性有重要的影响,可能是因为CaA、CA和FA都是羟基酚酸,只有在苯环上的官能团不同,CA有1 个羟基,CaA有2 个羟基,而FA有1 个羟基和1 个甲氧基。综上,酚酸衍生可提高果胶的抗氧化活性,提高的效率主要随酚酸结构、衍生程度(或总酚含量)及自由基种类而异,其次受果胶的结构特性影响。
2.6 胶提取物PFP及其衍生物的体外免疫活性分析
图5A显示,质量浓度0.2~1.0 mg/mL PFP及其衍生物处理后,RAW264.7细胞存活率大都维持在85%~110%,样品组间的差异不显著(>0.05),与0.001 mg/mL LPS处理组相比,除0.8~1.0 mg/mL PFP-L-CA处理组细胞存活率显著降低外,其他组比无显著变化(>0.05)。说明在所探讨的质量浓度范围内PFP果胶及其酚酸衍生物(0.8~1.0 mg/mL PFP-L-CA除外)对RAW264.7细胞无显著抑制作用(>0.05)。在RAW264.7细胞对中性红的吞噬能力方面(图5B),所有样品刺激的细胞吞噬能力都有浓度依赖性。与LPS处理组相比,质量浓度0.2~1.0 mg/mL PFP及0.2 mg/mL PFP-L处理会高度显著降低细胞吞噬能力(<0.001);而0.6~0.8 mg/mL PFP-L、0.4~1.0 mg/mL PFP-L-FA、0.4~1.0 mg/mL PFP-L-CA和0.8~1.0 mg/mL PFP-L-VC处理会显著促进细胞吞噬能力(<0.05、<0.001),酚酸(FA和CA)衍生组细胞吞噬能力最高达118%。在刺激RAW264.7细胞分泌NO方面(图5C),大部分样品刺激NO分泌量都有浓度依赖性。和空白组相比,0.2~1.0 mg/mL PFP处理组的NO生成量差异不显著(>0.05);0.2~0.6 mg/mL PFP-L、0.4~0.6 mg/mL PFP-L-FA、0.4~0.8 mg/mL PFP-L-CA和0.2~0.8 mg/mL PFP-L-VC处理组的NO生成量显著增加(<0.05、<0.01、<0.001),且PFP-L、PFP-L-FA和PFP-L-CA的刺激效果相似,在0.6~0.8 mg/mL质量浓度下刺激的NO生成量达最大(36~42 μmol/L),相当于0.001 mg/mL LPS组的72%~85%。说明过高剂量(质量浓度0.8~1.0 mg/mL)反而降低NO生成量,抑制免疫活性。
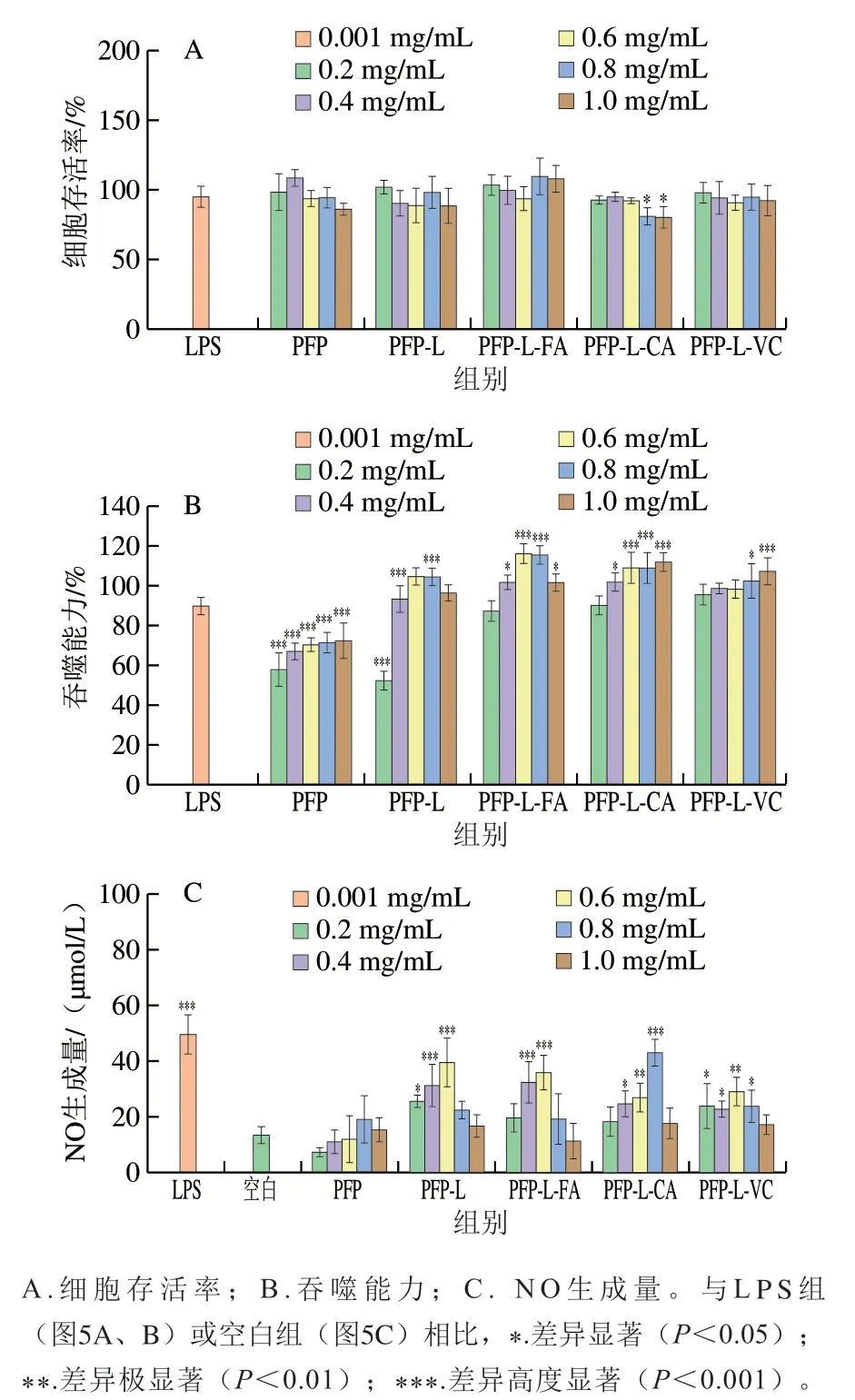
图5 小鼠巨噬细胞RAW264.7模型中果胶提取物PFP及其衍生物的免疫活性Fig. 5 Immunostimulatory activities of PFP and its derivatives in murine macrophage line RAW264.7
综上,添加漆酶与酚酸衍生的PFP具有显著增强体外免疫活性的作用,刺激RAW264.7细胞的吞噬和NO生成,酚酸(FA和CA)衍生物的活化作用比VC衍生组高。其免疫活性具有浓度依赖性,可能与酚酸和漆酶的存在有关,与样品的总酚含量(表1)和分子质量(表2)的变化无关。相比于具有免疫活性的RG-I区域组分类果胶或富含阿拉伯半乳聚糖的区域组分类多糖,果胶-酚酸衍生物因制备过程简单、成本低、绿色环保,并且仍具有果胶原本功能性(如凝胶、增稠、稳定性)等特点,故更具有研发与应用优势。
3 结 论
本研究主要分析了PFP及其CA与FA衍生物的分子特性和红外光谱差异,并研究了酚酸结构对PFP衍生物抗氧化活性和免疫活性的贡献差异关系。结果表明酚酸衍生可使PFP-L-FA和PFP-L-CA的总酚含量分别显著提高至11.07 mg/g和12.29 mg/g;分子性质方面,PFP-L-CA的提升至256.5 kDa,而PLP-L-FA的无明显变化(187.8 kDa),且两者的分子构象均由PFP的刚性短杆状变为链段较柔软的无规则线圈状;红外光谱显示衍生物分别在1 625 cm和1 739 cm的峰面积减少和增加均证明了酚酸通过酯键与PFP共价接枝,且衍生物粉粒的微观结构趋于松散和絮状化。此外,CA与FA衍生物的体外抗氧化活性(羟自由基、超氧阴离子自由基清除率和总还原力)和细胞免疫活性显著增强,体现在小鼠巨噬细胞NO生成量的增加和中性红吞噬能力的提升。综上,本研究所制备PFP酚酸(CA与FA)衍生物具有活性膳食纤维和保健食品的应用前景,研究为改性果胶的商业化应用提供理论参考,未来研究可详细解析衍生果胶与生物活性(如免疫和抗癌)的构效关系、机理、流变特性与加工稳定性。
