基于sigFeature方法的小麦不耐受患者差异蛋白筛选及通路研究*
2022-09-14哈尔滨医科大学卫生统计学教研室150081王玉鹏刘美娜
哈尔滨医科大学卫生统计学教研室(150081) 赵 敏 张 薇 孙 琳 王玉鹏 刘美娜
【提 要】 目的 应用蛋白质组学技术,基于sigFeature变量筛选方法,获得小麦不耐受患者血清差异表达蛋白;利用富集分析获得差异蛋白生物学解释,为小麦不耐受发病机制的研究提供依据。方法 收集小麦不耐受患者和对照组血清样本;应用TMT标记定量蛋白质组学技术获得蛋白表达数据、sigFeature方法筛选差异表达蛋白;进行差异蛋白GO功能注释和KEGG富集分析,外部数据集进行差异蛋白验证。结果 TMT技术鉴定蛋白849个,sigFeature筛选获得差异蛋白22个。富集分析结果:GO富集分析发现差异蛋白参与血小板脱颗粒、急性期反应等生物学过程;KEGG富集分析发现差异蛋白参与补体与凝血级联通路。外部验证结果:ITIH2蛋白的ROC 曲线下面积最大,AUC值为0.8673。结论 补体系统抑制和脂质代谢过程的改变是小麦不耐受发生的重要环节;ITIH2蛋白可能是小麦不耐受的关键蛋白;本研究从人体血清蛋白质组学的角度,为探究小麦不耐受的发生和调控机制提供依据。
近年来,食物不耐受流行程度大幅度增加,成为世界范围内的公共卫生问题[1];食物不耐受是指由于食物成分的化学作用,酶/运输缺陷等引起的变态反应性疾病,发生率约为15%~20%[2]。小麦不耐受在我国食物不耐受的种类中占有较高的比例[3-4],但发病机制尚不清楚,尚无从人体血清蛋白质组学方向的研究。作为蛋白质组学的重要分支,血清蛋白质组学可以利用质谱技术分析特定人群血清中的全部蛋白质,从整体水平上研究蛋白质的表达、结构、功能;结合统计学与生物信息学技术,能够获得差异蛋白、筛选生物标志物,为疾病发病机制的探索与研究提供依据[5]。
本研究从血清蛋白质组学角度入手,利用串联质谱定量法(tandem mass tag,TMT)对小麦不耐受患者与对照组血清进行蛋白质定量分析;利用sigFeature变量筛选方法获得差异蛋白,GO功能注释和KEGG富集分析解释差异蛋白生物学功能;并用外部数据集进行差异蛋白验证,为小麦不耐受发病机制研究提供方向和新思路。
资料与方法
1.研究对象
收集某医院变态反应科就诊患者血清,ELISA方法对人体血清中14种食物进行特异性IgG抗体检测;根据食物IgG水平将检测结果分为四个等级:<50U/ml为阴性,记作0级;50~100U/ml为轻度不耐受,记作+1级;100~200U/ml为中度不耐受,记作+2级;>200U/ml为重度不耐受,记作+3级[6];获得小麦不耐受患者及对照血清样本各7例,收集年龄、BMI、日常生活中食物摄入情况、家中是否养动物等基本信息。
2.蛋白质组学分析
TMT是用于标记不同蛋白样品并进行LC-MS/MS定量研究的体外标记技术。流程如下:①制备不同的蛋白样品;②Trypsin酶切获得相应样品的多肽;③利用不同的TMT标签标记样品;④等量混合成一个样品;⑤LC-MS/MS检测;⑥数据库检索与定量分析。
3.差异蛋白筛选
sigFeature是一种基于支持向量机-递归特征消除(support vector machine-recursive feature elimination,SVM-RFE)的特征选择算法[7]。SVM通过核函数将线性不可分数据映射到高维空间使其线性可分[8],尤其适用于小样本、高维数据分类模型构建[9];由于本研究特征数远大于样本数,因此选用线性核函数。SVM-RFE是基于SVM最大间隔原理的序列后向选择算法[10],该算法在迭代过程中剔除分类能力较弱的特征,保留分类能力较强的特征,迭代包括三个步骤:第一,训练SVM模型,获得每个特征的权重w;第二,根据特征排序标准计算每个特征得分;第三,剔除得分最小的特征,保留其余特征进行下一次迭代。待所有特征均被剔除后,根据特征剔除顺序获得特征重要性排序列表。sigFeature将SVM-RFE与t统计量结合,利用SVM计算权重,t统计量计算不同类别之间的差异,将两者结果的乘积作为特征排序标准,进行逐步特征选择。该方法能够更好地消除噪声和无关特征,筛选到数量较少、具有更高分类精度和良好生物学功能的特征。假设训练集{(x1,y1),…,(xn,yn),xj∈Rd,yi∈(-1,1)},xi={xi1,…,xid}是d维的输入向量,y是分类标签,则SVM的分类超平面f(x)可用方程式(1)表示,式中w=(w1,w2,…,wd)为权重,b为位移项。支持向量机权重的计算方式如公式(2)所示,αi为拉格朗日乘子。病例组与对照组的分离程度由方程式(3)表示,Δδ越大则两组间差异越大。n+、n-分别表示病例组和对照组样本数。
(1)
(2)
(3)
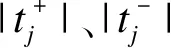
(4)
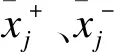
(5)
(6)
如果两组样本数均大于1,采用两独立样本t检验方法计算某一特征的组间差异,即公式(7)。由于本研究病例组和对照组样本数均大于1,因此,通过公式(7)计算特征的重要性排序得分。
(7)
4.数据分析
利用R软件获得差异蛋白,并进行GO功能注释和KEGG富集分析:GO功能注释分析差异蛋白参与的生物过程、分子功能、细胞组分;KEGG富集分析获得差异蛋白参与的最主要的代谢和信号转导途径。
5.外部验证
21例血清样本作为外部验证数据集,小麦不耐受患者7例,对照14例。绘制验证数据集ROC曲线并计算曲线下面积;AUC值作为衡量差异蛋白分类性能的指标,取值范围在0.5至1之间,值越大说明模型预测性能越好。
模拟实验
本研究利用模拟实验,比较sigFeature方法与随机森林(RF)、偏最小二乘法(PLS)在不同参数条件下对小样本蛋白质组学数据的变量筛选效果。模拟实验参数设置如下:总样本量设置为N=6,8,10,12,18,24,30,50,80,100,病例组与对照组样本量相等;总变量数为1000;差异变量比例为p=3%(30),5%(50),8%(80),10%(100);变量间相关性设置为r=0.2,0.4,0.6,0.8。比较三种方法在不同参数设置条件下的平衡准确度,以评价三种方法的变量筛选效果:平衡准确度=(灵敏度+特异度)/2。
模拟实验结果如图1~4所示。结果显示,不同相关系数条件下,样本量较小时,sigFeature方法变量筛选效果最好且较稳定,其次为PLS,RF较差且不稳定;随着样本量的增大,三种方法的平衡准确度均增大;当N=100时,sigFeature与PLS方法的平衡准确度趋近于1,高于RF。
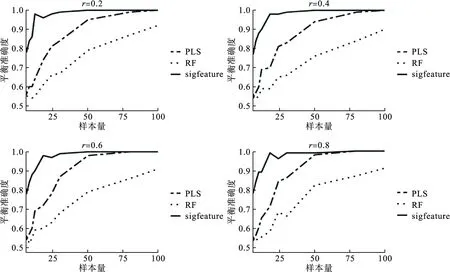
图1 p=3%(30)时,三种方法在不同相关系数下平衡准确度比较
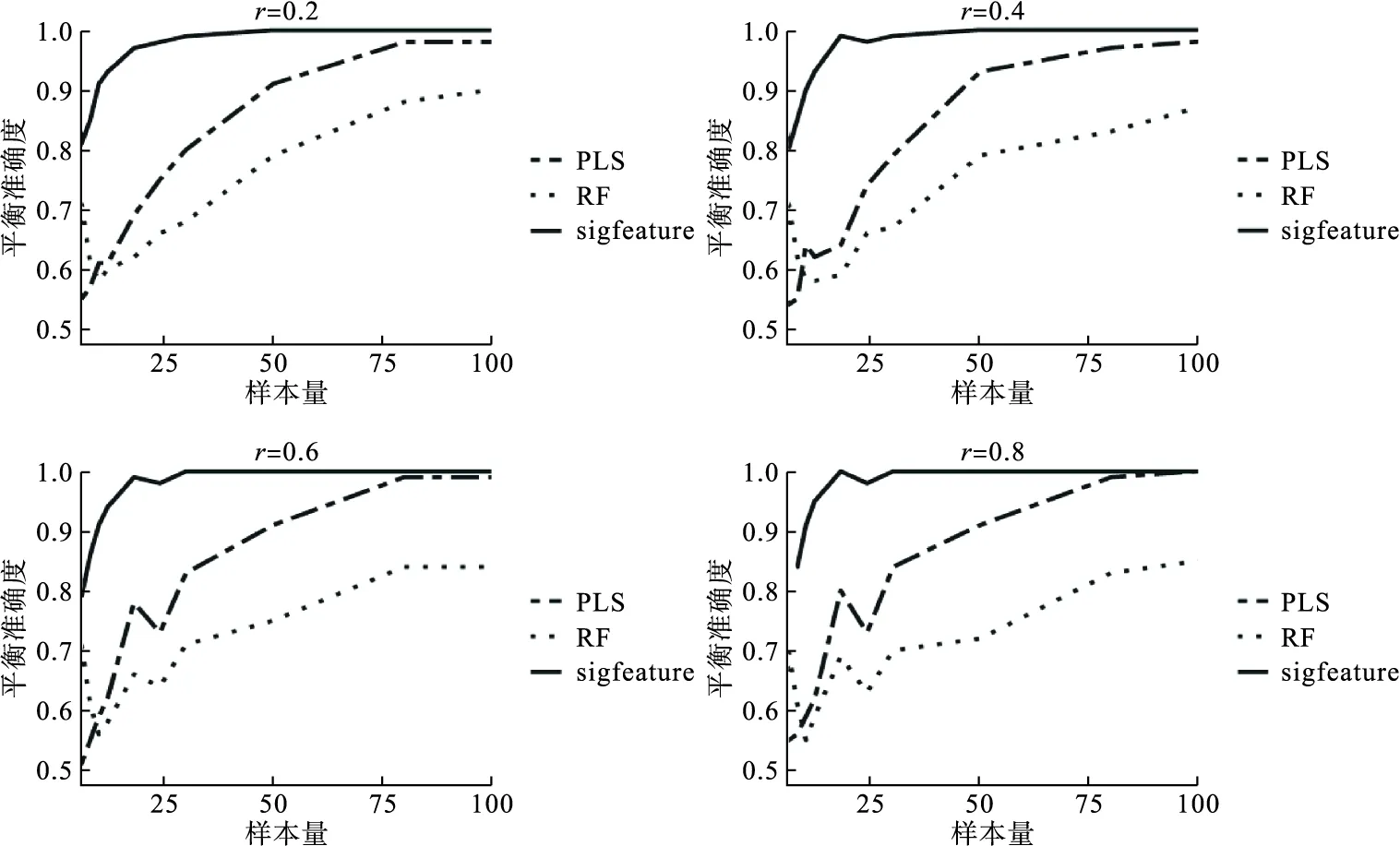
图2 p=5%(50)时,三种方法在不同相关系数下平衡准确度比较
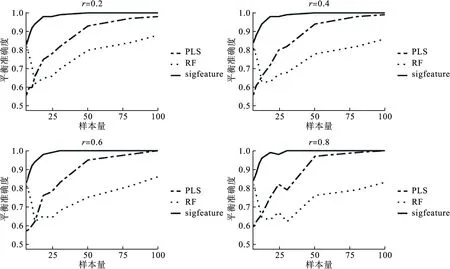
图3 p=8%(80)时,三种方法在不同相关系数下平衡准确度比较
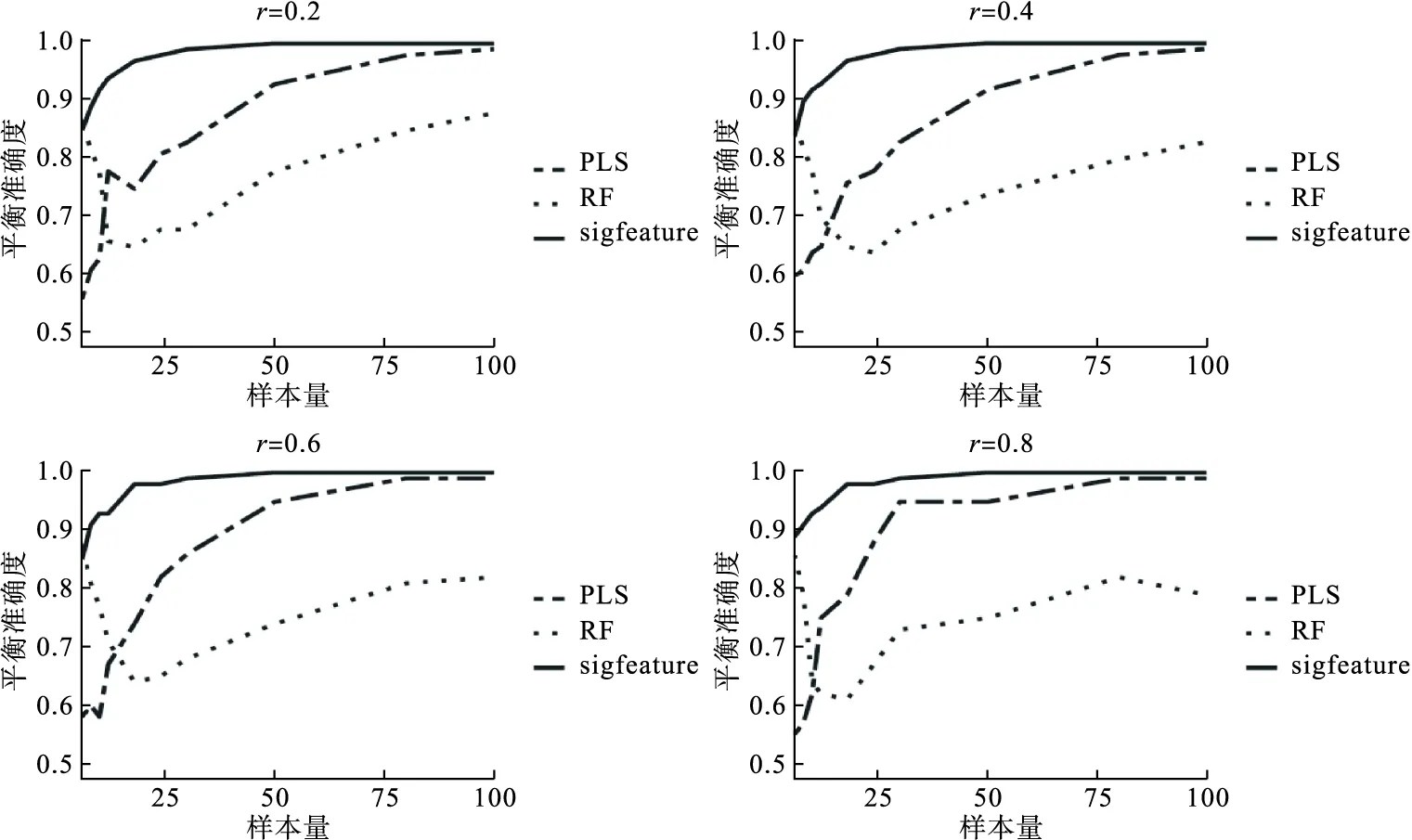
图4 p=10%(100)时,三种方法在不同相关系数下平衡准确度比较
结 果
1.基线资料比较
病例组与对照组单因素logistic回归分析结果见表1,年龄、BMI、水果、蔬菜、瘦肉及肝脏、海鲜、生冷食物、辛辣食物、维生素、是否养动物差异均无统计学意义(P>0.05),两组具有可比性,可以进行后续分析。
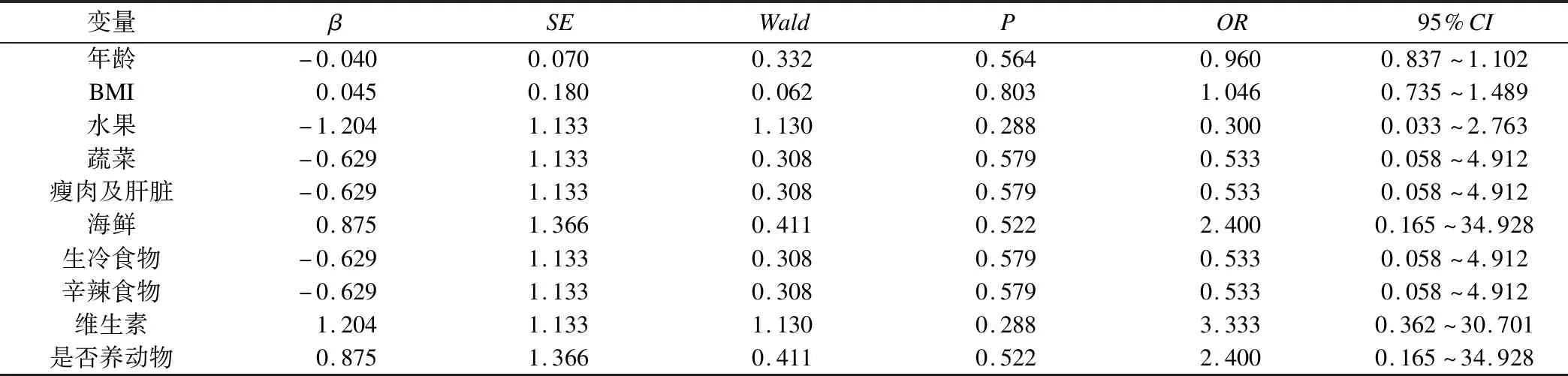
表1 小麦不耐受发生相关因素logistic回归分析结果
2.差异蛋白筛选
LC-MS/MS原始数据经过MaxQuant搜库和定量分析鉴定到849种蛋白质,其中上调465种,下调384种。sigFeature变量筛选获得蛋白重要性排序列表,排序靠前的22种蛋白对小麦不耐受患者和正常样本的分类效果最好,其中上调6种,下调16种,见表2。差异蛋白聚类分析结果的热图见图5,根据差异蛋白在不同组间的表达量可以将样本分为病例组和对照组,两组间无交叉。
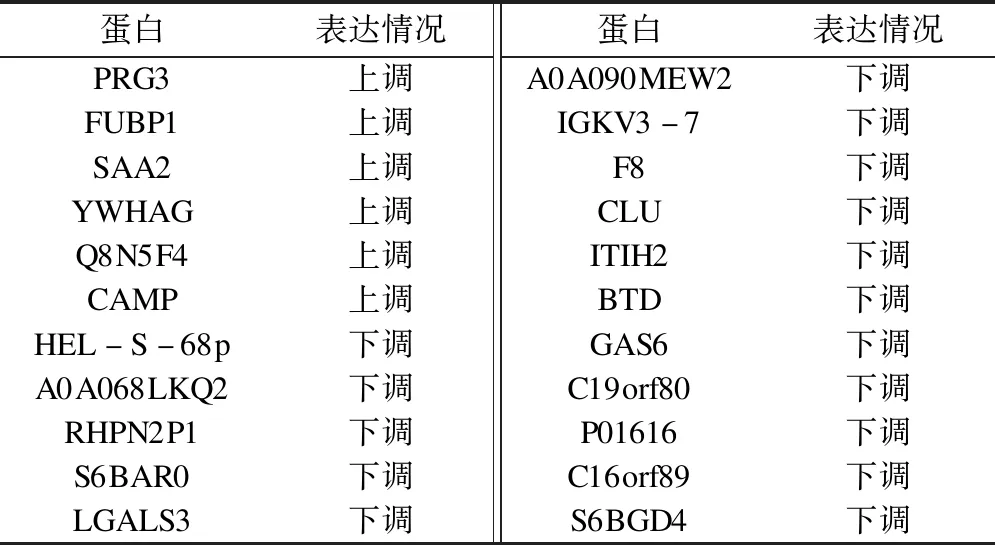
表2 差异表达蛋白
3.富集分析
GO富集分析结果见图6,差异蛋白参与的生物学过程主要集中在血小板脱颗粒过程、急性期反应过程、细胞蛋白质定位的正调控过程、细胞趋化性过程、白细胞凋亡调控等过程,参与的差异蛋白主要有F8、CLU、GAS6、SAA2、YWHAG、LGALS3、PRG3;细胞组成中,大部分蛋白位于分泌颗粒腔、细胞质囊泡腔、囊泡腔、血小板α颗粒管腔、血小板α颗粒等结构中,参与的差异蛋白主要有F8、CLU、GAS6、PRG3、CAMP、ITIH2、CLU、ITIH2、LGALS3、SAA2;差异蛋白分子功能主要是酶抑制剂活性、化学引物活性、受体酪氨酸激酶结合、蛋白酪氨酸激酶结合、蛋白激酶调节活性等,参与的差异蛋白主要有ITIH2、GAS6、YWHAG、LGALS3、SAA2、CLU、PRG3。KEGG富集分析发现差异蛋白富集在补体与凝血级联通路。
4.外部验证结果
sigFeature方法筛选获得22个差异蛋白,其中13个在验证数据集中没有表达,9个在验证数据集中表达,表达蛋白的AUC分布见表3。验证数据集ITIH2蛋白的AUC值最大,ROC曲线分析结果见图7,变量筛选数据集AUC为0.8163,曲线下面积95%置信区间为0.58~1,灵敏度为85.71%,特异度为71.43%;验证数据集AUC为0.8673,曲线下面积95%置信区间为0.67~1,灵敏度为85.71%,特异度为85.71%。
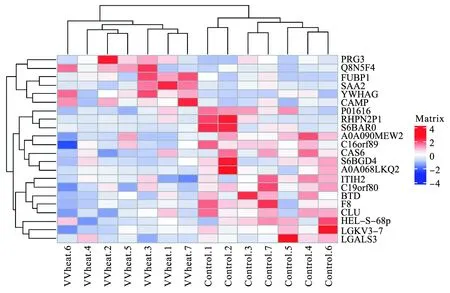
图5 差异表达蛋白聚类热图
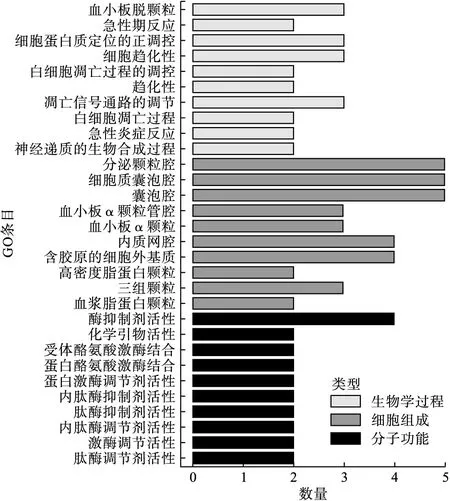
图6 差异蛋白GO富集分析结果
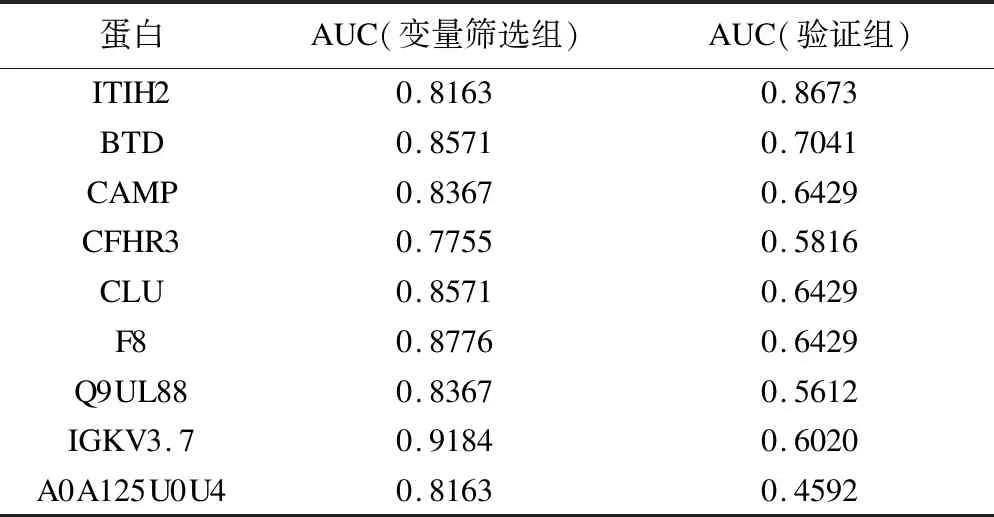
表3 变量筛选与验证数据集差异蛋白AUC分布
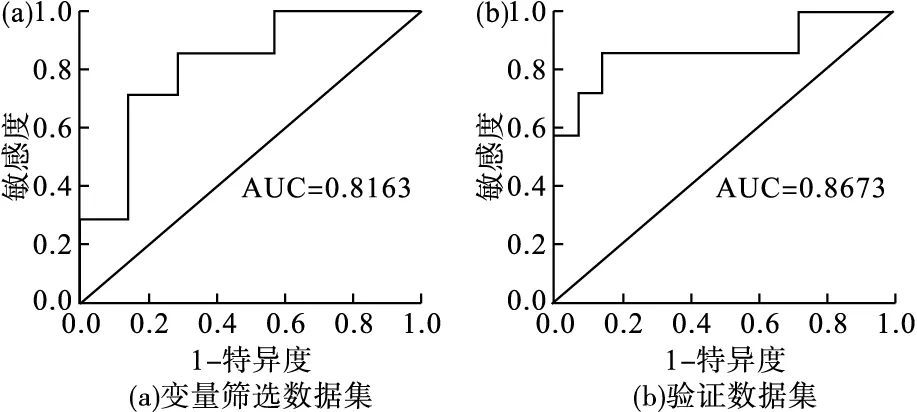
图7 ITIH2蛋白ROC曲线分析
讨 论
sigFeature是一种将SVM-RFE与t统计量结合的方法,具有适用于小样本高维组学数据,且能够筛选到分类精度更高的特征的优点,实际研究中主要用于基因表达数据的分类及变量筛选[7],本文将其用于小样本蛋白组学数据的研究中。模拟实验表明sigFeature方法在小样本蛋白质组学数据中具有较好的变量筛选效果,因此将其应用于小麦不耐受差异蛋白的筛选研究,但该方法计算相对复杂,运算速度较慢。
小麦中的某些蛋白质会使机体产应不耐受反应,引起消化、呼吸、皮肤、神经等多个系统的疾病。随着小麦年产量的增高,小麦不耐受人数也逐渐增加,因此,研究小麦不耐受致病机制及关键调控蛋白具有重要意义。目前关于小麦不耐受的研究较少且多从植物角度入手[11-13],有研究发现α-淀粉酶抑制剂、ω-5麦醇溶蛋白会引起小麦过敏反应及相关疾病;利用SDS聚丙烯酰胺凝胶电泳技术分析小麦蛋白成分,发现麦谷蛋白可能是引起小麦不耐受的蛋白。本研究从人体血清蛋白质组学角度入手,发现补体与凝血级联通路是差异蛋白主要参与的代谢通路,ITIH2蛋白是疾病调控的关键蛋白。
补体与凝血系统是先天性免疫的重要组成部分,主要由丝氨酸蛋白酶抑制剂及激活剂组成,通路激活后产生级联反应以抵御病原体入侵,促进伤口愈合,发挥机体的防御功能[14]。补体系统与凝血系统可通过多条途径相互作用,例如凝血因子XIIa可以激活补体因子C1r从而启动补体系统,凝血酶可以激活补体系统的C5释放C5a,C5a对中性粒细胞具有显著的趋化活性;相反,当抗凝系统被抑制时,补体通过增强血液凝固特性和增强炎症反应来促进血栓形成,进而增强凝血。补体和凝血级联通路的差异蛋白为CLU、F8。CLU为簇蛋白,是一种可溶性补体激活调节剂,能够调节末端补体级联反应并抑制C9与C5b-8复合物结合,调节促炎性细胞因子的生成,具有抑制补体系统和脂质转运的作用[15]。有研究发现CLU与鸡蛋过敏反应有关,但其机制需要进一步研究[16]。F8为凝血因子VIII,是血液凝固过程的关键蛋白,F8的缺乏会导致不同程度的出血紊乱[17]。有研究表明F8与免疫反应有关[18],本研究首次发现F8与小麦不耐受有关。
ITIH2为间α-胰蛋白酶抑制剂重链H2,由丝氨酸蛋白酶抑制剂组成。过去研究发现,α-淀粉酶/胰蛋白酶抑制剂是人类先天免疫反应的诱导剂,与小麦过敏反应有关,通过TLR4-MD2-CD14途径激活免疫细胞上的Toll样受体4,诱导抗原呈递细胞向外周淋巴结迁移,促进炎性趋化因子和细胞因子产生炎症反应,从而增强人体先天免疫作用[19-20]。本研究发现ITIH2蛋白在小麦不耐受的调控中发挥关键作用,外部验证中ITIH2蛋白的AUC值最高。
小麦不耐受的调控是一个复杂的过程,涉及到许多蛋白质共同参与,目前研究多为植物方向,无法阐明食物摄入后机体代谢过程的改变。因此,从人体血清蛋白质组学角度更容易理解疾病发生、发展的分子机制。本研究立足于人体血清蛋白质组学,发现补体和凝血系统的改变是小麦不耐受发生的重要环节,ITIH2蛋白是小麦不耐受的关键调控蛋白,为深入研究疾病的致病机制提供依据。
